Over the past two decades, research in the endothelin signaling system has been quite intensive since they are profoundly involved in hypertension and cardiovascular diseases. While major efforts are being invested in finding a proper antagonist to the two G-protein coupled receptors of endothelin, ET-A and ET-B, we provide a brief report on advances made in the field of the endothelin receptors and how Alomone Labs products have influenced the advances made.
Endothelins and Hypertension
Endothelin, a 21–amino acid peptide, has potent and sustained vasoconstrictive effects on vascular smooth muscle both in vivo and in vitro and is found in three different isoforms; ET-1, ET-2 and ET-3. Although expressed by other cell types, ET-1 is mainly expressed by the vascular system in endothelial cells. ET-1 binds and activates two G-protein coupled receptors (GPCRs), ET-A and ET-B (extensively reviewed in Reference 1). While the former has been attributed to vasoconstriction, the role of ET-B depends mostly on its location; on one hand it could promote vasoconstriction and on the other stimulate nitric oxide (NO) release-a known vasodilator2. ET-1 is implicated in pulmonary arterial hypertension as well as systemic hypertension models including deoxycorticosterone acetate-salt (DOCA) hypertension3-5. Contributing factors to the development of pulmonary hypertension are the elevated levels of circulating endothelin (ET-1). The direct implication of increased ET-1 on the pulmonary circulation was studied4. The study leads to the conclusion that following four weeks of chronic administration of ET-1, pulmonary hypertension was not developed. What was observed however, among other effects, following a 7-day administration of ET-1, was a three-fold increase of ET-B (but not ET-A) in both the intima and media of small pulmonary arteries detected under confocal microscopy imaging using Anti-ET-A and Anti-ET-B antibodies ((#AER-001 and #AER-002), respectively) which returned to normal after 28 days (Figure 1). Surprisingly, there was no increase in ET-1 expression in pulmonary arteries which was explained by the assumption that increased plasma ET-1 had no in vivo effect on vascular smooth muscle ET-1 production4 (Figure 1).
Chronic intermittent hypoxia (CIH) is characterized by very short hypoxic episodes followed by normoxia, and is the main distinguishing feature in sleep apnea which has been linked to hypertension in humans6. Some of the effects of CIH seem to occur in the carotid body (CB) where a 10-fold increase in ET-1 has been found in cats treated with CIH7. In the same study, the increased discharges observed in the carotid body of CIH-treated cats were decreased upon treatment with pharmacological blockers of ET-A and ET-B. Indeed, western blot analysis shows that ET-B is present in the carotid body and that its expression is increased in response to CIH. Immunohistological studies also show similar findings; that both receptors are present in glomus cells of the carotid body8 (Figure 2).
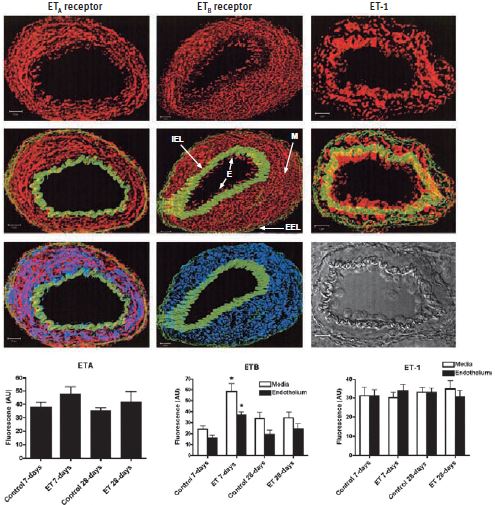
The images represent examples of composite Z stacks that were deconvolved and projected with the LSM 510 software (Zeiss). The first row of figures displays fluorescence for the ET-A and ET-B receptors, using Anti-ET-A and Anti-ET-B antibodies (#AER-001 and #AER-002) respectively and for ET-1, from left to right (in red). ET-B and not ET-A is detected in the endothelium (E). The second row displays the same components, but with the addition of the internal elastic lamina (IEL) and external elastic lamina (EEL; in green), which enables easy demarcation of the endothelium (E) from the media (M). The third row displays the fluorescence for smooth muscle actin (in blue), which is limited to the media (middle image) and colocalizes with ET-A (on the left). A phase contrast image is shown in the bottom right corner. Scale bar, 10 μm. The computed mean fluorescence intensities are presented in the bar graphs. * P < 0.01 versus control 7 days.
Adapted from reference 4 with permission of the American Thoracic Society.
Endothelins and Vascular Dysfunctions
The involvement of the endothelin signaling system is quite prominent and very much related to diabetes complications leading to vascular dysfunction in human and diabetic animal models9-12. Ca2+ channel blockers are clinically widely used for the management of hypertension, angina pectoris, and cardiac arrhythmias13. The long term effects of benidipine (a Ca2+ channel blocker) on the endothelin system were studied in a rat model of human Type 2 diabetes14. This model shows an increase in ET-1 expression in plasma and left ventricular (LV) tissues compared with the non-diabetic controls. The diabetic rat model also shows increased levels of the ET-A and ET-B receptors as demonstrated by immunohistological studies as well as western blot analysis using Alomone Labs’ Anti-ET-A and Anti-ET-B (Figures 3A and B). Upon an 8 week treatment with benidipine, ET-1, ET-A and ET-B levels were similar to non-diabetic controls. In addition, benidipine treatment strongly prevented cardiomyocyte hypertrophy, partly attributed to the decrease in expression levels of ET-1 and its related molecules14.
Endothelins and the Renal System
The endothelin receptor system is distinctly involved in the regulation of renal function under both physiological and pathophysiological conditions. The effects of ET-1 in kidney have been attributed to both subtypes of GPCRs by the use of specific agonists and antagonists of these receptors. The receptors also have functions leading to vasoconstriction as well as vasodilation, important in the regulation of renal vascular resistance15,16. However, direct evidence for the cellular distribution of both receptors was limited until recently. Using Anti-ET-A, immunohistochemistry studies showed that ET-A colocalizes with desmin in mesangial cells17 (Figure 4A). In the renal cortex, ET-A also is found to colocalize with aquaporin 2 (using Anti-Aquaporin 2 antibody (#AQP-002)) in collecting ducts but not in the outer medulla. In order to indisputably show that the signals were specific to the receptor, the immnunoreactivity of the ET-A receptor was completely abolished by preabsorbing Anti-ET-A with the antigen used for immunization17 (Figure 4B).
The dopaminergic system also controls the renal system. In a recent paper, it was shown that D3 dopamine receptors work in close concert with ET-B in maintaining proper Na+ homeostasis in the kidney. In fact, these two receptors colocalize in immunohistochemistry studies18 (Figure 5).
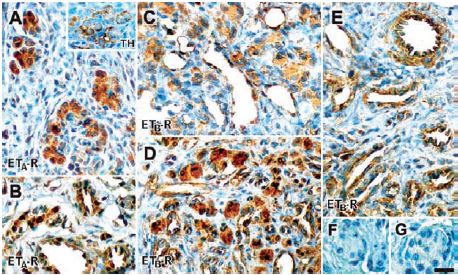
A) Using Anti-ET-A antibody (#AER-001), ET-A immunoreactivity is located in cells within the carotid body lobules; these cells are also positive for TH (inset). B) Positive ET-A staining in arterioles and venules in the carotid body vascular pole. C) and D) ET-B immunoreactivity using Anti-ET-B antibody (#AER-002), was shown in the glomus cell cytoplasm, endothelium of the carotid body parenchymatous capillaries and venules. E) ET-B immunoreactivity was also shown in arteries, endothelium and tunica media of the vessels in the vascular pole. ET-B immunoreactivity was more intense in glomus cells of the CIH-exposed carotid bodies (D) compared with control carotid bodies (C). F) and G) Negative controls preabsorbed with control peptides showed no staining. Scale bar, 20 μm.
Adapted from reference 8 with permission of the Histochemical Society.
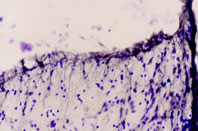
Immunohistochemical staining of rat dorsal peri-ventricular thalamus using Anti-ET-B antibody (#AER-002). Cresyl violet is used as the counterstained. The specific staining is black.
1. Anti-ET-B antibody (#AER-002), (1:200).
2. Anti-ET-B antibody, preincubated with the negative control antigen.
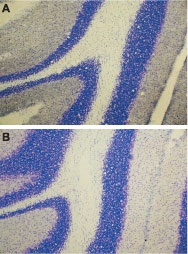
Immunohistochemical staining of rat cerebellum using Anti-ET-A antibody (#AER-001) (A), or Anti-ET-A antibody preincubated with the negative control antigen (B). Cresyl violet is used as the counterstained. The specific staining is black.

1. Anti-ET-A antibody (#AER-001), (1:200).2. Anti-ET-A antibody, preincubated with the negative control antigen.
Endothelins, Retina and Neurons
In most cases, photoreceptor injury leads to the activation of Muller cells in the retina, where the accumulation of glial fibrillary acidic protein (GFAP) is observed. Although this type of cellular response has probably evolved to limit neuronal damage, excessive activation of Muller cells can impair recovery19,20. The activation of Muller cells in response to diverse retinal disorders suggests that these cells are at the health checkpoint of retinal neurons and perhaps do so by sensing a neuron-derived signaling molecule released by damaged or healthy neurons21. Briefly, this possibility was assessed by using mouse retina subjected by a severe light insult (consequently leading to retinal degeneration) and monitoring changes in gene expression21. Microarray analysis was performed on retinas damaged by acute exposure to visible light (known to also lead to photoreceptor death). Genes that were upregulated include ET-2, which accumulated in photoreceptors, and ET-B which accumulated in the inner nuclear layer (INL) where Muller cells are normally distributed. Immunostaining using Anti-ET-B and anti-GFAP showed that the localization of these two proteins overlapped (Figure 6). These observations suggest that ET-2 may function as a stress signal by activating ET-B located on Muller cells, thereby activating these cells21. In contrast, ET-1 seems to be the activator of both ET-A and ET-B receptors (found in bipolar dendrites and horizontal cells, using Anti-ET-A and Anti-ETB respectively) following a non-acute light insult, where a similar cell survival mechanism takes place22.
An additional indication that the endothelin signaling system is present and functional in the neuronal system is from a recent study where ischemia was inflicted in mice and ET-B presence was monitored among various other parameters. ET-B colocalized with GFAP and vimentin around the ischemic lesion in immunohistological studies while no receptor was detected in GFAP-/- Vim-/- mice suggesting that the intermediate filaments, GFAP and vimentin, are required for the production, stability, or distribution of ET-B in reactive astrocytes23(Figure 7). In addition, using cultured astrocytes the same study showed that by laser-scanning confocal microscopy, there was a remarkable colocalization between bundles of intermediate filaments and ET-B immunoreactivity in wild type cells23 (Figure 8).
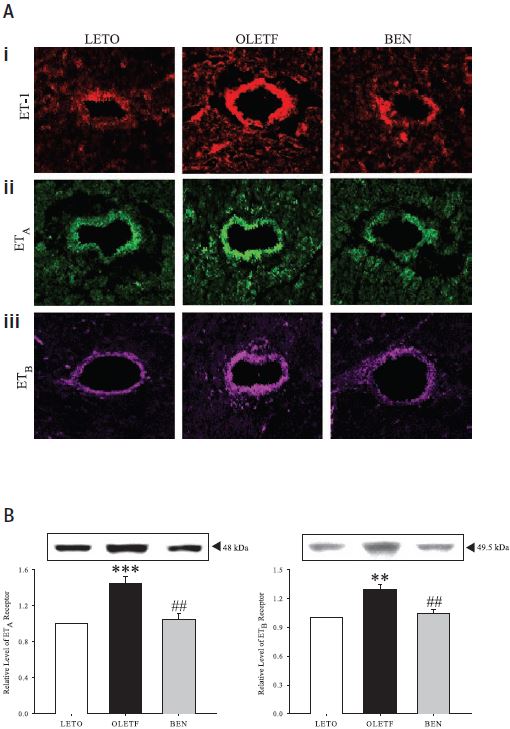
A) Confocal images showing showing immunofluorescence labeling for ET-1 (i), ET-A (ii) and ET-B (iii) in left ventricular tissue sections from control rats (LETO), diabetic rats (OLETF) and benidipine treated diabetic rats (BEN) using Anti-ET-A and Anti-ET-B antibodies respectively (#AER-001 and #AER-002). Both vascular and extravascular regions are more immunoreactive in diabetic rats than in those in control rats and Benidipinetreated rats. Original magnification is x400. B) Western blot analysis of ET-A (left) and ET-B (right) from left ventricular tissues of control, diabetic and benidipine-treated rats. ± SE; n = 12 animals. **P < 0.01 and ***P < 0.001, compared with corresponding values obtained in LETO rats; #P < 0.01, compared with corresponding values in OLETF rats without any treatment.
Adapted from reference 14 with permission of the American Physiological Society.
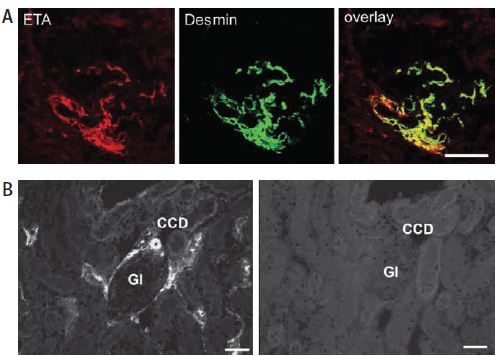
A) Using Anti-ET-A antibody (#AER-001), ET-A receptor immunoreactivity colocalizes with desmin on extra- and intraglomerular mesangial cells as evidenced by the yellow color in the overlay picture. B) Preabsorbed with the peptide used for immunization, no ET-A immunoreactivity can be observed (right panel). CCD, cortical collecting ducts; Gl, glomerulus.
Adapted from reference 17 with permission of the Histochemical Society.
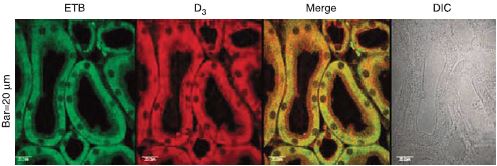
D3 dopamine receptor and ET-B colocalize using Anti-ET-B antibody (#AER-002) in kidneys from rat. Colocalization appears as yellow after merging the images of fluorescein isothiocyanate-tagged ET-B receptor (green) and Alexa 568-tagged D3 receptor (red). No staining is seen without the antibodies (DIC). DIC, differential interference contrast.
Adapted from reference 18 with permission of Nature Publishing Group.
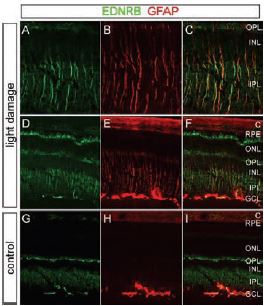
A)-F) Mice were exposed to 6 hours of bright light with pupil dilation, followed by a 24 hour recovery in darkness. A)-C) Enlarged views of the central retina from D)-F). G)-I) Control mice maintained in darkness. Light exposure leads to the accumulation of ET-B as shown using Anti-ET-B antibody (#AER-002) and anti-GFAP in Muller cells, characterized in the central retina by their radial fibers. ET-B also accumulates in presumptive astrocytes in the GCL and at the outer tips of the Muller cells, which lie alongside the photoreceptor inner segments between the outer nuclear layer and the RPE. Among Muller cells in the light-damaged retina, both ET-B and GFAP show substantial cell-to-cell variability. C, Choroid; ONL, outer nuclear layer; OPL, outer plexiform layer; IPL, inner plexiform layer.
Adapted from reference 21 with permission of The Society for Neuroscience.

A) and B) No difference is detected in the distribution of astrocytes in the penumbra in the two groups although astrocytes of GFAP -/-Vim -/- mice show less prominent hypertrophy of cellular processes. Green, S100β; blue, cell nuclei visualized by ToPro-3. Using Anti ET-B (#AER-002), reactive astrocytes adjacent to the ischemic lesion in WT mice (visualized by antibodies against GFAP, C and I) were ET-B positive (E and K) 7 days later after MCA transection. F) and L) In contrast, no ET-B was detected in astrocytes in GFAP -/- Vim -/- mice. G) and M) Merged images of (C and E) and of (I and K) respectively. C)-N) Red, GFAP; green, ET-B; blue, nuclei visualized by ToPro-3. Scale bar, 50 μm.
Adapted from reference 23 with permission of Nature Publishing Group.
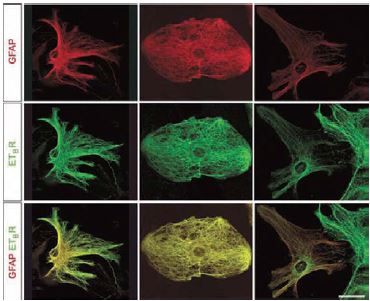
Laser-scanning confocal microscopy reveals a filamentous appearance of ET-B immunostaining using Alomone Labs’ Anti-ET-B (#AER-002), which colocalizes with GFAP-positive bundles of intermediate filaments. Red, GFAP; green, ET-B. Scale bar, 20 μm.
Adapted from reference 23 with permission of Nature Publishing Group.
References
- Rubany, G.M. and Polokoff, M.A. (1994) Pharmacol. Rev. 46, 325.
- Tykocki, N.R. et al. (2009) J. Pharmacol. Exp. Ther. 329, 875.
- Dai, X. et al. (2004) Hypertension 43, 1048.
- Migneault, A. et al. (2005) Am. J. Respir. Crit. Care Med. 171, 506.
- Sauvageau, S. et al. (2006) Exp. Biol. Med. 231, 840.
- Quan, S.F. and Gersh, B.J. (2004) Circulation 109, 951.
- Rey, S. et al. (2006) Brain. Res. 1086, 152.
- Rey, D. et al. (2007) J. Histochem. Cytochem. 55, 167.
- Ferri, C. et al. (1997) Diabetologia 40, 100.
- Piatti, P. et al. (1996) Diabetes 45, 316.
- Sachidanandam, K. et al. (2008) Am. J. Physiol. 294, H2743.
- Sachidanandam, K. et al. (2009) Am. J. Physiol. 296, R952.
- Roberts, R. and Zanchetti, A. (1996) Cardiovascular Drug Therapy.
- Jesmin, S. et al. (2006) Am. J. Physiol. 290, H2146.
- Lanese, D.M. and Conger, J.D. (1993) J. Clin. Invest. 91, 2144.
- Hyslop, S. and de Nucci, G. (1992) Pharmacol. Res. 26, 223.
- Wendel, M. et al. (2006) J. Histochem. Cytochem. 54, 1193.
- Zeng, C. et al. (2008) Kidney Int. 74, 750.
- Bringmann, A. and Reichenbach, A. (2001) Front. Biosci. 6, 72.
- Guidry, C. (2005) Prog. Retin. Eyes Res. 24, 75.
- Rattner, A. and Nathans, J. (2005) J. Neuroscience 25, 4540.
- Torbidoni, V. et al. (2006) Exp. Biol. Med. 231, 1095.
- Li, L. et al. (2008) J. Cereb. Blood Flow Metab. 28, 468.