KCNEs are integral membrane proteins that associate with and strongly modulate the activity of several ion channels. The different KCNE isoforms are widely and differentially expressed in muscular and neuronal tissues as well as in epithelial cells. Mutations in KCNE genes were shown to lead to disruptions of diverse physiological systems and diseases such as cardiac arrhythmias, deafness and paralysis. It is becoming increasingly apparent that KCNE auxiliary subunits are capable of interacting with many different ion channels. These interactions are the scope of this brief review.
Introduction
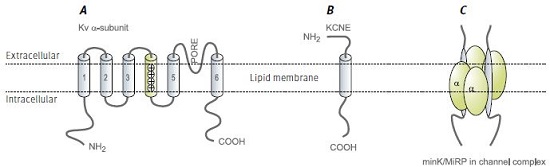
Functional voltage dependent K+ channels are constructed primarily by tetramerization of KV α subunit pore forming proteins (Figure 1). With about 40 genes encoding KV channel subunits, which may ensemble by either homo or hetero-tetramerization, the molecular and functional diversity of native voltage dependent K+ currents is huge1 .
In addition, several groups of proteins referred to as auxiliary subunits were shown to interact and modify KV currents. These include KVβ, KChiP and KCNE proteins. As each of these consists of several isoforms and each pore forming tetramer may interact with different combinations of auxiliary subunits, further KV current heterogeneity is derived from interactions with auxiliary subunits1,2.
KV channels and auxiliary subunits participate in the regulation of many cellular processes that form the basis of normal functioning of literally every physiological system. These range from regulation of the heartbeat and neuronal coding to fluid and salt secretion in epithelial tissues such as the kidney and inner ear. KV channels were also implicated as key regulators of cell fate, either as an obligatory element in the progression of apoptosis3 or as a control element in cell cycle progression and cellular proliferation4,5.
KCNE Subunits Structure and Naming
KCNE are membrane-spanning proteins that interact with ion channels and modify their properties. Their structure consists of a single membrane-spanning α helix, and extra- and intracellular N and C termini, respectively (Figure 1B). Five human genes were identified and denoted KCNE1-5. The origin of this name stems from the realization that KCNE proteins are KV channel auxiliary subunits and therefore these proteins are part of the KV channel superfamily (all human genes of this family begin with KCN). The first isoform to be characterized was KCNE1 (IsK), which was originally named MinK standing for minimal K+ channel. Subsequently, the related isoforms were named Mink Related Peptides or MiRPs2.
Physiological Relevance of KCNE Subunits
Several mutations in KCNE genes lead to disease. Long QTs (LQT) are syndromes in which cardiac arrhythmias may occur due to prolongation of the cardiac action potential. This cellular phenomenon is, in many cases, a result of reduction in the magnitude of the K+ efflux, which facilitates repolarization of the cell membrane and action potential termination. In addition to inherited mutations in pore forming KV channels, mutations in KCNE1 and 2 were found in patients with these cardiac syndromes. Such observations emphasize the importance of KCNE auxiliary subunits in the overall regulation of K+ homeostasis and the cellular mechanisms that depend on it. This is further illustrated by other mutations, causing deafness (KCNE1) or disturbed muscle function (KCNE3)2.
The KCNE auxiliary subunits regulate KV channel activities in many ways, including enhancement or depression of permeation properties and change in gating characteristics. However, in many cases the modulatory action of KCNE on KV channels is complex, and may combine an impact on the pore and voltage sensing properties2.
Knockout of the KCNE1 gene in mice resulted in phenotype alterations that hint to the involvement of these gene products in cellular volume regulation in epithelial cells6, a task that is common to many K+ channels and is also related to apoptosis3.

Immunohistochemical staining of KCNE2 (MIRP1) in mouse heart, using Anti-KCNE2 (MIRP1) Antibody (#APC-054). Immunoreactivity (red) appeared in striate pattern in heart muscle.
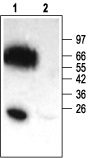
1. Anti-KCNE2 (MiRP1) Antibody (#APC-054) (1:200).
2. Anti-KCNE2 Antibody, preincubated with the negative control antigen.
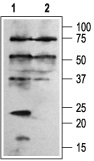
1. Anti-KCNE1 (IsK) Antibody (#APC-163) (1:200).
2. Anti-KCNE1 Antibody, preincubated with the control fusion protein.
KCNE Subunit Interactions
By using ion channel toxins and antibodies, the stoichiometry of KV channel interaction with KCNE subunits was defined as 2 KCNE per functional channel (i.e. a KV tetramer)7 (Figure 1C). In addition, studies pointed out that the KCNE C-terminus (intracellular) interacts with the pore region of the KV α subunit8,9.
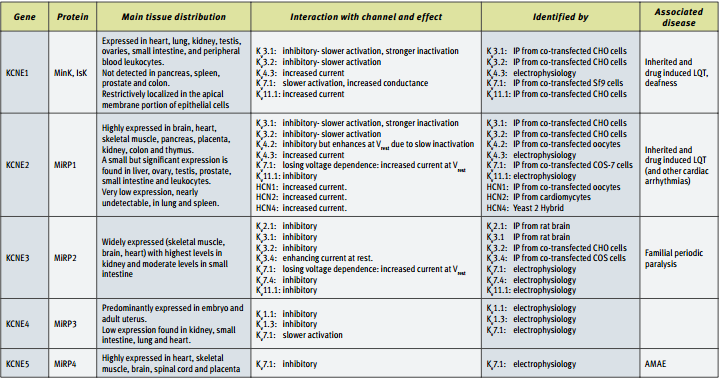
Originally, MinK (KCNE1) was shown to associate with KV7.1 (KCNQ1 or KVLQT), and this channel’s subunit composition was suggested as corresponding for part of the delayed rectifier K+ current in cardiac tissue10,11. Indeed, as mentioned before, mutations in both genes result in very similar pathological symptoms (i.e. LQT)1. Subsequently, other known KCNE isoforms, MiRP1 and MiRP2 were shown to modulate other components of the cardiac and skeletal K+ current. MiRP1 (KCNE2) associates and suppresses the other cardiac delayed rectifier component carried out via KV11.1 (HERG) channels12, as well as the transient (A type) component associated with KV4.213.
MiRP2 (KCNE3) enhances the activity of the skeletal muscle KV3.4 channel. Mutations in this gene were found in some patients with familial periodic paralysis. It was suggested that disrupted KCNE3 causes reduced K+ currents at resting membrane potential, leading to membrane depolarization. Interestingly, it was shown that this association reduced the ability of a peptidyl toxin BDS II to block the KV3.4 currents, hence, demonstrating that the pharmacology of native channel complexes may differ from that of the cloned pore forming α subunit14.
It is interesting to note that while KCNE1 endows KV7.1 with clear, slow voltage dependent activation10,11 (similar results were obtained with KCNE415 and KCNE516), the association of these pore-forming channels with KCNE217 and KCNE318, results in currents that lack voltage dependency and may serve as “leak” K+channels. Regarding the specificity of the interactions it should be noted that KCNE5 was found to modulate KV7.1 currents with no major influence on other KV7 combinations or on KV11.1 channels16.
In experimental expression systems, KCNE proteins were shown to interact with and influence a number of KV and related channels (See Table). The physiological significance of these reported interactions is not always clear and not all of these interactions were detected in vivo. In a number of studies, KCNE2 was implicated as an auxiliary subunit of the KV related HCN channels19-21. Hyperpolarization activated channels (HCN) are similar in structure to the archetypal KV channel. However, while all other voltage dependent channels tend to open in response to membrane potential depolarization, HCN channels open upon membrane potential hyperpolarization. In turn, channel activation results in a net cation influx, which depolarizes the membrane potential, back to rest and beyond. These characteristics equip the cells that express HCN channels with the molecular machinery that is needed for pacing the membrane potential. Hence, the KCNE2 isoform plays a critical role in the generation of cells with pacing capabilities, such as the S-A node in the heart and in pacemaker neurons. It is important to note that the other KCNE isoforms had no such an effect on HCN4 channels20.
KCNE proteins are expressed in the brain where KCNE3 was shown to associate with and inhibit the K+currents carried via KV2.1 and KV3.1 channels22. These results are in line with the observation that KV3.1 (and KV3.2) currents are slowed down once the pore-forming gene is co-expressed with KCNE1, KCNE2 or KCNE323. When tested for its modulatory activity on KV1 channels (the Shaker subfamily), KCNE4 was identified as an inhibitory subunit of KV1.1 and KV1.3 channels. It had no apparent effect on other KV1 channels, suggesting that KCNE4 channels specifically interact with these two KV isoforms24.
KCNE subunits were shown to be an important factor in determining the excitability characteristics and capabilities of cardiac-, skeletal-, smooth-25 muscle and neuronal cells. The patterns of protein associations described above, highlight the necessity of fine-tuning in K+ handling for “proper” physiological functioning. Recently, the expression of several KCNE isoforms was demonstrated in several uterine cancer cell lines26. In addition, mice carrying a truncation mutation in KV7.1 channel, the major known partner of KCNE, had increased susceptibility to gastric cancer development27. Such observations may stress the role played by ion channels in general and KCNE auxiliary subunits in particular, in the cellular mechanisms leading to cancer.
Acknowledgments
We thank Dr. B. Attalli from The Tel Aviv University for his comments.
IP = immunoprecipitation.
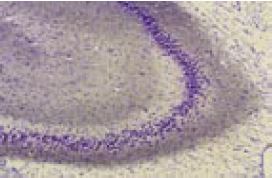
Immunohistochemical staining of rat hippocampus with Anti-KV4.2 Antibody (#APC-023). Note that the molecular layers were stained while the mossy fiber terminal region was not stained.
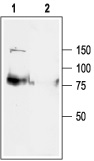
1. Anti-KCNQ1 Antibody (#APC-022) (1:200).2. Anti-KCNQ1 Antibody preincubated with the negative control antigen.
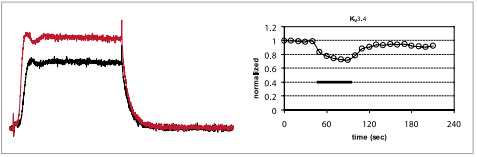
Inhibition of KV3.4 channels expressed in Xenopus oocytes. Left: example traces before (red) and during (black) bath perfusion of 1 μM BDS-II (#B-450). Holding potential was –100 mV, test potential to 0 mV (100 ms) was delivered every 10 seconds. Recordings were made while perfusing ND96 Buffer. Right: time course for the experiment shown on the left. The vertical bar indicates the period of BDS-II perfusion.
References
- Yu, F. H. and Catterall, W. A. (2004) Sci. STKE. 2004, re 15.
- Abbott, G. W. and Goldstein, S. A. N. (2001) Mol. Interven. 1, 95.
- Meir, A. (2003) Modulator 18, 2.
- Bronstein-Sitton, N. (2002) Modulator 17, 4.
- Bogin, O. (2003) Modulator 18, 24.
- Lock, H. and Valverde, M. A. (2000) J. Biol. Chem. 275, 34849.
- Chen, H. et al. (2003) Neuron 40, 15.
- Melman, Y. F. et al. (2004) Neuron 42, 927.
- Romey, G. et al. (1997) J. Biol. Chem. 272, 16713.
- Barhanin, J. et al. (1996) Nature 384, 78.
- Sanguinetti, M. C. et al. (1996) Nature 384, 80.
- Smith, P. L. et al. (1996) Nature 379, 833.
- Zhang, M. et al. (2001) Circ. Res. 88, 1012.
- Abbott, G. W. et al. (2001) Cell 104, 217.
- Teng, S. et al. (2003) Biochim. Biophys. Res. Commun. 303, 808.
- Angelo, K. et al. (2002) Biophys. J. 83, 1997.
- Tinel, N. et al. (2000) EMBO J. 19, 6326.
- Schroeder, B. C. et al. (2000) Nature 403, 196.
- Yu, H. et al. (2001) Circ. Res. 88, e84.
- Decher, N. et al. (2003) Pflugers Arch. Eur. J. Physiol. 446, 633.
- Qu, J. et al. (2004) J. Biol. Chem. 279, 43497.
- McCrossan, Z. A. et al. (2003) J. Neurosci. 23, 8077.
- Lewis, A. et al. (2004) J. Biol. Chem. 279, 7884.
- Grunnet, M. et al. (2003) Biophys. J. 85, 1525.
- Ohya, S. et al. (2002) Am. J. Physiol. 282, G277.
- Suzuki, T. and Takimoto, K. (2004) Int. J. Oncol. 25, 153.
- Elso, C. M. et al. (2004) Hum. Mol. Genet. 13, 2813.