Maintaining glucose homeostasis is an important factor in mammalian physiology, and is controlled to a large extent by the pancreas. Specialized beta cells residing in the Islets of Langerhans sense the blood glucose levels and secrete insulin, which signals these levels to the rest of the body. Ion channels play a major role in many aspects of this cellular process and serve as targets for pharmacological interventions that treat diseases in which glucose homeostasis is impaired. In type-2 diabetes, the tight link between glucose sensing and insulin secretion is impaired due to mutations (as well as other genetic and acquired changes) in a specific K+ channel and most medications for this condition target this channel. This short review describes the mechanisms underlying the physiology and pathophysiology of insulin secretion with emphasis on the role ion channels play in controlling and modulating it.
The Pancreas
The main function of the pancreas, a gland attached to the intestine, is to control blood glucose levels. However, most of the pancreas is comprised of exocrine tissue, which secretes enzymes into the intestine, while only about 1% of the tissue consists of endocrine cells, which are clustered in the Islets of Langerhans.
Most of the endocrine cells in the islet are called beta cells. These respond to an increase in the plasma glucose level by secreting insulin into the blood circulation, which in turn allows cells (mainly in muscle and liver) to absorb glucose. (For more information see the website mentioned in Ref. 1)
Other cell types in the pancreas further control the plasma glucose levels. Alpha cells secrete glucagon, which stimulates mitochondrial respiration in target cells, when the glucose level is low. Delta cells secrete somatostatin onto the islet, which inhibits both insulin and glucagon secretion. Lambda cells secrete GLP-1, which causes an increase in glucagon and insulin secretion. All these cell types contribute to synchronization in the “whole islet” response.
Cells in the Islet of Langerhans are Excitable
Like neurons, pancreatic endocrine cells respond to their relevant stimulation with plasma membrane depolarization (caused by changes in the activity of ion channels) and action potential firing, which leads to vesicle exocytosis (controlled by elevation of Ca2+ concentration, mediated by ion channels) and hormone secretion.2
Thus, glucose stimulation leads to depolarization, which is often oscillatory and is accompanied by oscillations in Ca2+ and NADH concentrations and in mitochondrial membrane potential, often leading to oscillatory insulin secretion. Ion channels play a major role in a network of cellular and molecular feedback mechanisms that produce these dynamics.2-7
Resting Potential and Initiation of Hormone Secretion
The mechanism that links glucose sensing to hormone secretion has been extensively studied in beta cells and involves the activity of several ion channels at key points in the cascade (for detailed reviews see Refs. 2, 8-9). K+ channels that are sensitive to ATP are plasma membrane protein complexes composed of four Kir6.2 (KCNJ11) pore-forming subunits surrounded by four SUR1 (sulphanylurea receptor, of the ABC transporter protein family) auxiliary subunits. These protein complexes sense the amount of glucose entering a beta cell since the activity of KATP channels depends on the amount of ATP in the cytoplasm, which in turn depends on the amount of glucose absorbed by the beta cell. The activity of KATP channels is negatively correlated to the amount of ATP, i.e. increases in ATP levels tend to reduce the activity of the channel.9
KATP channels are the main channels that are open during resting conditions and therefore their activity sets the resting membrane potential. Closure of KATP channels by increased ATP concentration leads to membrane depolarization, which causes opening of voltage dependent Ca2+ (CaV) channels, leading to Ca2+ influx.2 The exact molecular make-up of CaV channels in beta cells probably differs between species and is still somewhat controversial.10 However, the main CaV channels that control insulin secretion are L-type channels of the CaV1 subfamily (CaV1.2 and/or CaV1.3).11-13 In addition, using a CaV1.3 knockout mouse it was demonstrated that this channel is essential for the normal development of the pancreas.14 Other CaV channels, members of the CaV2 (P/Q, N and R types) channel subfamily have been detected in these cells and in several cases their functional contribution was demonstrated.15-17 Members of the CaV3 (T-type) channels were also detected in some beta cells preparations.10
The mechanism of coupling between depolarization and secretion is very similar in beta cells and in neuronal synaptic transmission. Further emphasis on the major role played by CaV1 channels in insulin secretion is given by demonstrations of physical and functional interaction of CaV channels with synaptic proteins (such as syntaxins and SNAP-25) that control vesicle exocytosis.18-20
The above-mentioned channels and pathways comprise the principle route for transduction of the blood glucose level into blood insulin signal (Fig. 1). However, many more channels are involved in fine-tuning and regulation of this signal transduction pathway and these are briefly described below.
Voltage dependent K+ (KV) channels also sense the depolarization initiated by KATP closure and their delayed activation leads to repolarization and termination of exocytosis. In beta cells, KV2.1 channels were demonstrated to be the main KV channel that mediates this termination,21-26 which in part is based on their interactions with synaptic proteins.27,28 Other KV channels (see below) are expressed in beta and other endocrine cells but their exact functional role is still not fully resolved.23,26
Ca2+ activated K+ channels open in response to intracellular Ca2+ ([Ca2+]i ) elevation and in turn cause repolarization of the membrane potential, often leading to attenuation of the [Ca2+]i signal. Via such a feedback loop, KCa2 and/or KCa3 channels (of the small/intermediate conductance protein family) contribute to oscillations in membrane potential and [Ca2+]i .29,30 The presence of KCa1.1 (large conductance, BK, Slo) channels was confirmed but they did not contribute to glucose induced electrical activity.31
Several Kir3 (GIRK or G protein activated inward rectifier K+ channels) which are structurally similar to the Kir6.2 subunit of KATP , were detected in islet cells. Kir3 (mainly Kir3.4 but also Kir3.2, GIRK4 and GIRK2) channels were found in alpha as well as beta cells. These G-protein activated K+ channels contribute to hyper and/or repolarization of the membrane potential, once they are opened. Their exact role is not completely resolved, but they probably contribute to reduction in insulin release, in response to hormones and transmitters such as Ach, epinephrine and somatostatin.32-35
In addition, a few K2P channels have been detected in the whole pancreas at the RNA level (PCR). K2P5.1 (KCNK5, TASK-2) was found to be expressed in the islet as well as exocrine tissue, and K2P10.1 (TREK-2) is expressed in the insulinoma cell line MIN6.36
TRP channels are sensitive to various stimuli and their activation modulates both [Ca2+]i and membrane potential, therefore contributing to regulation of secretion. The TRPM5 channel, which is permeable to monovalent cations and gated by internal Ca2+ is expressed in the pancreas, where it might contribute to membrane potential oscillations.37 Recently, the involvement of the Ca2+ permeable TRPM2 in insulin secretion was reported. It involves ADP ribose activation of this channel as well as temperature dependence.38The activation by ADP ribose might be related to oscillations in NADH concentration that have been reported to occur in beta cells in response to glucose activation. In addition, two splice variants of TRPC4 which were cloned from mouse insulinoma cell lines have been suggested to act as heteromeric (with other TRPC) Ca2+channels.39,40
Intracellular RyR and/or IP3R channels on the ER membrane contribute to shaping Ca2+ signals and therefore, insulin secretion.7,41 These channels release Ca2+ from ER into the cytoplasm and are linked to [Ca2+]i oscillations possibly via interaction with TRP channels.41
Some evidence suggests that islet cells may under certain conditions (low glucose for example) respond to ATP (which may have been coreleased with insulin) via P2X channels.42 Such a mechanism would further increase secretion, as these ATP receptors respond to ATP binding by opening a Ca2+ permeable pore. However, expression of P2 receptors in the pancreas is developmentally regulated, and for example, P2X7 (in alpha cells) is expressed only in adults and might be related to aging.43 This might be in correlation to the suggested role for this channel in supporting apoptosis.

Repertoire of Ion Channels in Different Types of Endocrine Cells
Alpha cells respond to glucose by decreasing glucagon secretion. This might be achieved by glucose acting on beta cells to release GABA, which in turn activates GABA(A)R on alpha cells and causes inhibition of glucagon secretion. However, once the glucose level is low, GABA is not released from beta cells and alpha cells go on secreting glucagon.44 Another proposed mechanism involves insulin secreted from beta cells, which reduces the alpha cell KATP channels’ sensitivity to ATP inhibition. This means that glucose in now less effective in inducing glucagon secretion from alpha cells.45
In comprehensive studies that have addressed this issue, alpha cells are reported to express the following channels: KV3.1, the silent subunits KV6.1 and possibly KV9.2.26 At the level of a single cell RT-PCR and electrophysiology experiments have suggested the following Ca2+ and Na+ channels as mediators of voltage dependent inward currents in alpha cells: CaV1.2 and CaV1.3 (L-type), CaV2.1 (P/Q-type), CaV2.3 (R-type) and NaV1.7 (TTX-sensitive).17
Beta cells respond to increase in glucose concentration by insulin secretion. Beta cells are reported to express the following KV channels: KV2.1, KV3.2 and the silent subunits KV6.2 and KV9.3.26 Reports that KV11.1 (erg1) is also expressed and is involved in glucose induced insulin release are based on PCR and some pharmacology.46 Shaker type KV1 channels were found along with KV2 channels, but these probably do not contribute to secretion.23 RT-PCR and electrophysiology experiments at the level of the single cell suggested that the following Ca2+ and Na+ channels mediate voltage dependent inward currents in beta cells: CaV1.2 (L-type), CaV2.1 (P/Q-type), CaV2.2 (N-type), CaV2.3 (Rtype) and NaV1.7 (TTX-sensitive).17
Delta cells secrete somatostatin, which inhibits both alpha and beta cell secretion. One possible mechanism for this inhibitory action is via inhibition of oxidative respiration in beta cells.47 Another proposed mechanism is the activation of Kir3 channels by somatostin receptors on alpha and beta cells.33 Delta cells are reported to express KV2.2 and possibly the silent KV9.2.26 Another report implies that the KV3.4 channel acts as the major delayed rectifier and shows electrical activity following glucose stimulation and NaV (TTX-sensitive) as well as CaV channel activity.48 Lambda cells secrete GLP-1, which promotes glucagon and insulin secretion. GLP-1 probably acts on beta cells via GPCR dependent mechanisms, which involves elevation of cAMP that increases the L-type CaV current and inhibits KV channels.49
Use of Specific Channel Modulators and Neurotoxins in the Study of Endocrine Physiology
KATP channel blockade leads to insulin secretion while openers of KATP block insulin secretion; both approaches are used clinically to tackle diabetes and hyperinsulinism, respectively. Extensive reviews cover this important topic and it will not be discussed here in detail.8,50,51
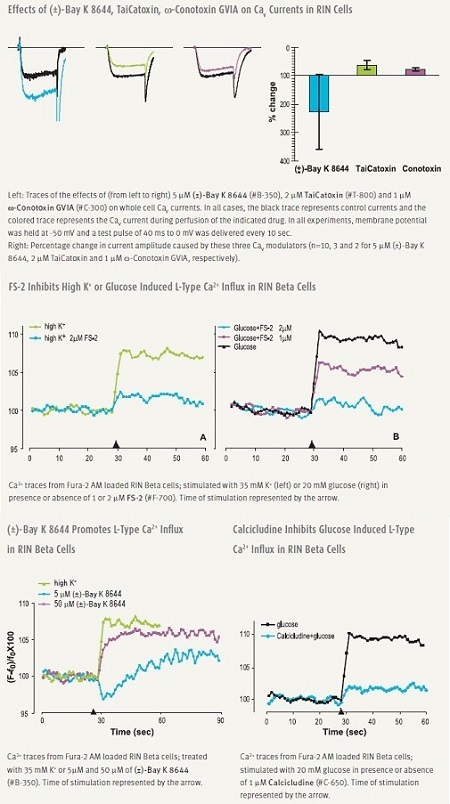
Pharmacological manipulation of CaV1 (L-type) channels modulate secretion. DHPs (DiHydroPyridines) are widely used, relatively specific L-type modulators. (±)-Bay K8644 is a potent enhancer of L-type current and is often used to induce [Ca2+]i elevations and insulin secretion.12 Several snake venom toxins, TaiCatoxin, Calcicludine, Calciseptine and FS-2 are specific and potent inhibitors of L-type channels, some of which have been shown to be effective in beta cells (Figure 2).52
As mentioned before, the main CaV channel involved in insulin secretion is one of the CaV1 family members. However, although other CaV channels have been detected in beta cells, their role is not fully resolved. The role played by these channels might be to regulate and/or fine tune the release process. A nice example is illustrated by an experiment in which SNX-482, a specific CaV2.3 channel blocker, was shown to inhibit only the second phase of insulin release.16
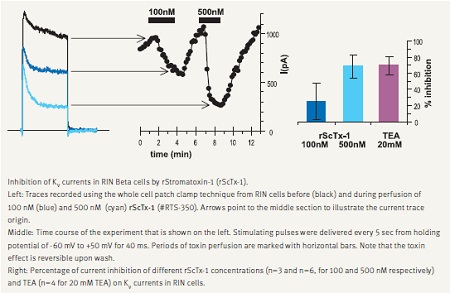
In accordance with its major role in depolarization and termination of exocytosis, KV2.1 channel blockade increases secretion. This was demonstrated with a few spider toxins such as Hanatoxin and Guangxitoxin, which are specific blockers of this channel.21,22 We demonstrate that Stromatoxin-1 (ScTx-1) is a potent inhibitor of KV currents recorded in RIN insulinoma cells (Fig. 3).
Synaptic neurotoxins have been used to affect secretion. α-Latrotoxin application induced both ion channel modulation (in a direction favoring increased exocytosis) as well as direct enhancement of insulin containing vesicle exocytosis.52,53 We demonstrate that α-Latrotoxin potently affects Ca2+ dependent enzyme phosphorylation in RIN cells (Fig. 4).
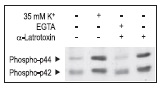
α-Latrotoxin promotes activation of p42/44 MAP Kinase by inducing Ca2+ influx in RIN insulinoma cells. Cells were treated for 15 min with 35 mM K+ or 2 nM α-Latrotoxin (#LSP-130) in presence or absence of 5mM EGTA. The picture presents Western blot analysis of active p42/p44 MAP Kinase probed with a phospho p42/p44 MAPK antibody.
Channelopathies Related to Diabetes
Mutations and polymorphism in KATP channel components cause a few congenital as well as acquired conditions (via increased susceptibility).8,50,51
Gain of function mutations usually make KATP less sensitive to ATP blockage, resulting in increased channel activity, decreased insulin release and diabetes.
Loss of function mutations either eliminate KATP channels from the plasma membrane or increase their sensitivity to ATP block. This causes decreased channel activity, leading to increased insulin secretion, hyperinsulinism and hypoglycemia. This depolarized beta cell state may eventually lead to apoptosis, beta cell death and diabetes.
Recently two gain of function mutations in CaV1.2 (Timothy syndrome)54 and in NaV1.7 (familial erythermalgia)55 were described, as these two channels are expressed in islet cells, mutations in their coding might influence the normal functioning of the pancreas.
For humans, the consequences of glucose imbalance might be severe. Currently the principle way to tackle such conditions in the clinic is with pharmacophores that bind to KATP and shift their ATP sensitivity. However, many ion channels participate in the control of secretion in pancreatic endocrine cells and therefore, in the maintenance of glucose homeostasis. Therefore, the fact that the modulation of secretion can be fine tuned renders it amenable to many possible interventions.
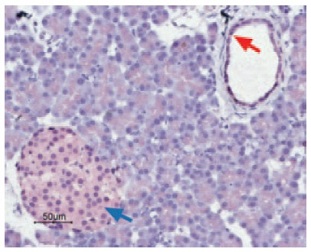
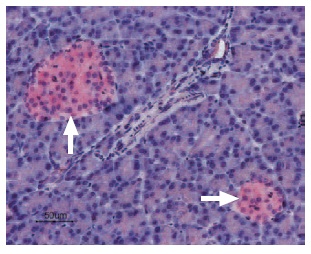
Immunohistochemical staining of CaV1.2 in rat pancreas was visualized using Anti-Human CaV1.2 (CACNA1C) Antibody (#ACC-022) (1:50). Paraffin embedded sections of rat pancreas showing both endocrine (Islets of Langerhans) and exocrine areas. Strong and highly specific staining is evident in both endocrine cells (blue arrow) and arteries endothelium (red arrow). Universal Immunoalkaline- phospathase Polymer followed by New Fuchsin Substrate (Histofine, Nichirei corp) was used for the color reaction. Counterstain is Hematoxilin.
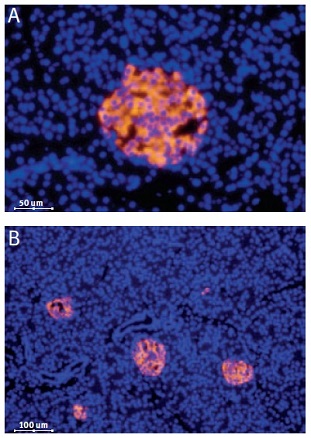
Immunofluorescent staining of CaV1.3 in rat pancreas was visualized using Anti-CaV1.3 (CACNA1D) Antibody (#ACC-005) (1:50). Paraffin embedded sections of rat pancreas showing both endocrine (Islets of Langerhans) and exocrine areas. Strong and highly specific staining is evident only in endocrine cells (red). Secondary antibody is goat antirabbit Alexa 555 (1:400). Counterstain is Hoechst 33342 (blue). (A) 200x (B) 100x.

Immunohistochemical staining of Kir3.4 in rat pancreas was visualized using Anti-KCNJ5 (Kir3.4) Antibody (#APC-027) (1:50). Paraffin embedded sections of rat pancreas showing both endocrine (Islets of Langerhans) and exocrine areas. Strong and highly specific staining is evident only in endocrine cells (arrow). Universal Immuno-alkaline-phospathase Polymer followed by New Fuchsin Substrate (Histofine, Nichirei corp) was used for the color reaction. Counterstain is Hematoxilin.
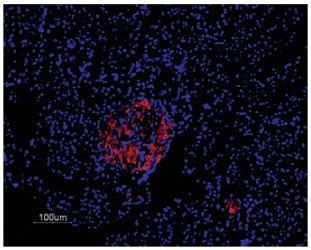
Immunofluorescent staining of KV2.1 in rat pancreas was visualized using Anti-KV2.1 Antibody (#APC-012) (1:50). Paraffin embedded sections of rat pancreas showing both endocrine (Islets of Langerhans) and exocrine areas. Strong and highly specific staining is evident only in endocrine cells (red). Secondary antibody is goat anti-rabbit Alexa 555 (1:400). Counterstain is Hoechst 33342 (blue).
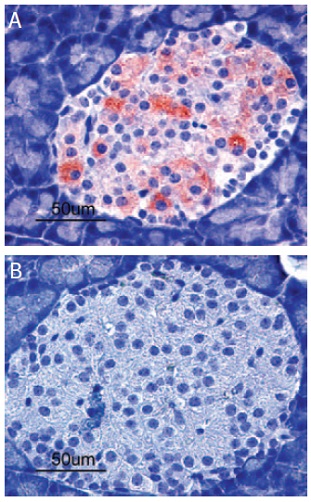
Immunohistochemical staining of Kir6.2 in rat pancreas. Paraffin sections of rat pancreas were visualized by Anti-Kir6.2 Antibody (#APC-020) (1:100). (A) Strong granular staining in a number of cells within the Islets of Langerhans is readily detected (red). (B) The negative control slide shows no staining.
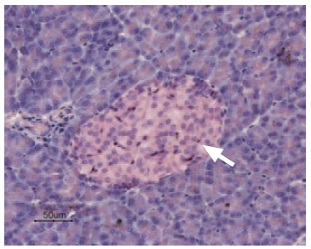
Immunohistochemical staining of KCNK5 in rat pancreas was visualized using Anti-KCNK5 (TASK-2) Antibody (#APC-037) (1:50). Paraffin embedded sections of rat pancreas showing both endocrine (Islets of Langerhans) and exocrine areas. Strong and highly specific staining is evident only in endocrine cells (arrow). Universal Immuno-alkaline-phospathase Polymer followed by New Fuchsin Substrate (Histofine, Nichirei corp) was used for the color reaction. Counterstain is Hematoxilin.
References
- Thomson, A.B.R. and Shaffer E.A. (2000 update) First Principles of Gastroenterology.
- MacDonald, P.E. and Rorsman, P. (2006) PLoS Biol. 4, e49.
- Berggren, P.O. et al. (2004) Cell 119, 273.
- Rolland, J.F. et al. (2002) Diabetes 51, 376.
- Gilon, P. et al. (1999) J. Biol. Chem. 274, 20197.
- Luciani, D.S. et al. (2006) J. Physiol. 572, 379.
- Tamarina, N.A. et al. (2005) Diabetes 54, 3073.
- Ashcroft, F.M. (2005) J. Clin. Invest. 115, 2047.
- Nichols, C.G. (2006) Nature 440, 470.
- Yang, S.N. and Berggren, P.O. (2005) Am. J. Physiol. Endocrinol. Metab. 288, E16.
- Rosario, L.M. et al. (1993) Pflugers Arch. 424, 439.
- Barg, S. et al. (2001) Biophys. J. 81, 3308.
- Liu, G. et al. (2004) Mol. Pharmacol. 65, 1269.
- Namkung, Y. et al. (2001) J. Clin. Invest. 108, 1015.
- Pereverzev, A. et al. (2002) Mol. Endocrinol. 16, 884.
- Jing, X. et al. (2005) J. Clin. Invest. 115, 146.
- Vignali, S. et al. (2006) J. Physiol. 572, 691.
- Yang, S.N. et al. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 10164.
- Cho, W.J. et al. (2005) J. Cell. Mol. Med. 9, 380.
- Kang, Y. et al. (2002) Diabetologia 45, 231.
- Herrington, J. et al. (2006) Diabetes 55, 1034.
- Herrington, J. et al. (2005) J. Physiol. 567, 159.
- MacDonald, P.E. et al. (2001) Mol. Endocrinol. 15, 1423.
- Tamarina, N.A. et al. (2005) Am. J. Physiol. Endocrinol. Metab. 289, E578.
- MacDonald, P.E. et al. (2002) J. Biol. Chem. 277, 44938.
- Yan, L. et al. (2004) Diabetes 53, 597.
- MacDonald, P.E. et al. (2002) Mol. Endocrinol. 16, 2452.
- Leung, Y.M. et al. (2003) J. Biol. Chem. 278, 17532.
- Zhang, M. et al. (2005) J. Gen. Physiol. 126, 353.
- Tamarina, N.A. et al. (2003) Diabetes 52, 2000.
- Li, Z.W. et al. (1999) J. Neurophysiol. 81, 611.
- Vaughn, J. et al. (2000) Biochem. Biophys. Res. Commun. 274, 302.
- Smith, P.A. et al. (2001) J. Physiol. 532, 127.
- Ferrer, J. et al. (1995) J. Biol. Chem. 270, 26086.
- Yoshimoto, Y. et al. (1999) FEBS Lett. 444, 265.
- Duprat, F. et al. (2005) J. Physiol. 562, 235.
- Prawitt, D. et al. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 15166.
- Togashi, K. et al. (2006) EMBO J. 25, 1804.
- Qian, F. et al. (2002) Diabetes 51, S183.
- Cruz-Cruz, R. et al. (2005) Am. J. Physiol. Endocrinol. Metab. 289, E439.
- Gustafsson, A.J. et al. (2004) FASEB J. 19, 301.
- Petit, P. et al. (1998) Bri. J. Pharmacol. 125, 1368.
- Coutinho-Silva, R. et al. (2001) Cell Tissue Res. 306, 373.
- Wendt, A. et al. (2004) Diabetes 53, 1038.
- Leung, Y.M. et al. (2006) Endocrinology 147, 2155.
- Rosati, B. et al. (2000) FASEB. J. 14, 2601.
- Daunt, M. et al. (2006) Endocrinology 147, 1527.
- Gopel, S.O. et al. (2000) J. Physiol. 528, 497.
- MacDonald, P.E. et al. (2002) Diabetes 51, S443.
- Ashcroft, F.M. and Rorsman, P. (2004) Rev. Endocr. Metabo. Disord. 5, 135.
- Farret, A. et al. (2005) Fundam. Clin. Pharmacol. 19, 647.
- Lajus, S. et al. (2006)J. Biol. Chem. 281, 5522.
- Silva, A.M. et al. (2005) J. Physiol. 565, 783.
- Splawski, I. et al. (2004) Cell 119, 19.
- Cummins, T.R. et al. (2004) J. Neurosci. 24, 8232.