The G-protein coupled receptors (GPCRs) are the largest and most versatile protein family in the mammalian genome. They interact with G-proteins to activate many intracellular signaling pathways and modulate ion channel activity. GPCRs modulate ion channels by two distinct pathways. The main focus of this review is the direct modulation of N-type, P/Q-type Ca2+ and Kir3 K+ channels by the Gβγ subunit, the membrane delimited pathway. Since both ion channels and GPCRs play an important role in intracellular signaling, they have become targets in the search for therapeutic drugs for cancer, heart diseases, obesity and others.
The GPCRs Superfamily
Intracellular signaling, the cellular tool for communication, is a very complex and diverse process. This process relies on elaborate systems of proteins which enable cells to respond to a variety of stimuli in their internal or external environment.1,2 Among these membrane proteins is the GPCRs Superfamily. They form the largest and most diverse family of cell surface receptors and proteins. For example, there are about 1000 GPCRs in neurons alone. The diversity of this superfamily is a result of the large number of members comprising this family, their ability to form different dimer combinations and their ability to respond to a multitude of stimuli, as well as by the large number of intracellular signaling pathways they activate.2,3
Despite their structural and functional diversity, all GPCRs share a similar molecular architecture. They consist of seven transmembrane domains, linked by alternating intracellular and extracellular loops, an extracellular N-terminus and an intracellular C-terminus.3
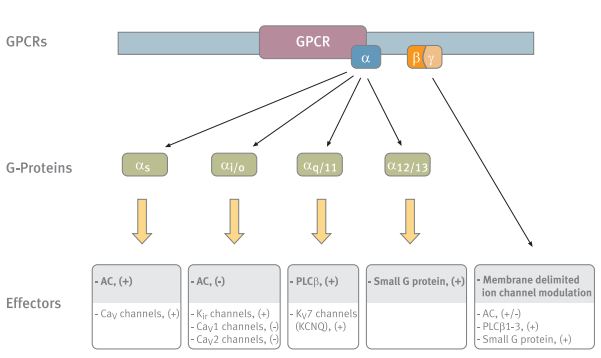
The different G-protein subunits and their various effectors. The main effectors of each subunit are shaded. Abbreviations: AC- adenylyl cyclase, PLC-β- phospholipase C-β.
An enormous amount of signal molecules, hormones, neurotransmitters, chemokines and local mediators, can activate different GPCRs with high specificity. GPCRs are associated with distinct classes of heterotrimeric GTP-binding proteins (G-proteins).2-5
The G-proteins consists of three subunits: α, β and γ. To date, about 17 genes encoding the α-subunits, 5 for β-subunits and 12 for the γ- subunits, have been identified.5,6 The interaction of G-proteins with GPCRs leads to the dissociation of the Gα subunit from the Gβγ, and either subunit may act as a downstream effector. The Gβγ acts as a dimer subunit, enhancing the receptor mediated signal by activating a distinct and diverse array of end targets such as enzymes and ion channels.5-7
These signaling events are terminated following the reassociation of the G-protein heterotrimer. This is due to the intrinsic GTPase activity of the Gα subunit, which hydrolyzes bound GTP to GDP. This activity was shown to be accelerated by a group of proteins, GTPase-activating proteins that were named regulators of G-protein-Signaling (RGS). The RGS exert their action by interacting directly with the Gα subunit.8,9

Immunohistochemical staining of CaV2.1 channel with Anti-CACNA1A (CaV2.1) Antibody (#ACC-001) (1:100) in mouse cerebellum. (A) CaV2.1 channel (red) appears in Purkinje cells (horizontal arrows) and is distributed diffusely in the molecular layer (Mol) including in astrocytic fibers (vertical arrows). (B) staining of astrocytic fibers with glial fibrillary acidic protein (blue – originally green digitally edited to blue) in the section demonstrates the location of astrocytic fibers in the molecular layer. (C) Confocal merge CaV2.1 and GFAP.
The G-proteins have been classified into four major categories based on the sequence similarity of their Gα subunits: Gαs, Gαi/o, Gαq/11 and Gα12/13. (Fig.1) This classification defines both receptor specificity, as well as, in most cases, effector specificity.5,6 The Gs and Gq have two well-defined effector pathways, the adenylyl cyclase (AC) and the phospholipase C-β (PLCβ) pathway, respectively.6 The activation pathways of the Gi and Go are much less defined.

Immunohistochemical staining of CaV2.2 channel with Anti-CACNA1B (CaV2.2) Antibody (#ACC-002) (1:100) in mouse cerebellum. (A) CaV2.2 channel (red) appears in Purkinje cells (arrows) and is distributed diffusely in the molecular layer (Mol). (B) staining of Purkinje nerve cells with mouse anti-calcium binding protein antibody (green) demonstrates the restriction of CaV2.2 to cell bodies but not to dendrites in the molecular layer. (C) Confocal merge of CaV2.2 and CBD 28K.
For many years researchers focused on the role that the different Gα subunits play in GPCRs activation pathways. However, today it is well established that the Gβγ subunit also mediates signal transduction by interacting with different effectors including AC, PI3 kinase, proteins of the MAPK pathway, and several ion channels.5,6 (Fig. 4)
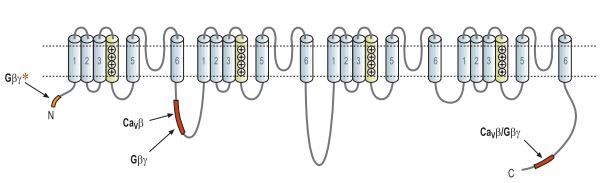
Putative Gβγ binding site.
Ion channels are essential for the function of excitable cells by mediating electrical currents and controlling specific ion concentrations; however, they are also widely expressed in non-excitable cells.
Ion channels comprise a large family of transmembrane proteins. They regulate the movement of ions across the cellular membrane and can be divided into groups according to their ion specificity: Na+ channels, Ca2+channels, K+ channels, Cl– channels and nonspecific cation channels.10,11 Most ion channels are multisubunit proteins that undergo post-translational modification processes.12
GPCRs can modulate ion channel activity through two distinct pathways:
1. An indirect pathway that involves a common second messenger leading to the phosphorylation of the channel.
2. A direct pathway, involving binding of Gβγ directly to the channel also known as membrane delimited modulation.13-16
A mechanism has been proposed which tries to explain the striking specificity of the signaling pathways, in spite of GPCRs diversity and the diversity of their effectors. This mechanism suggests the existence of a pre-assembled macromolecular signaling complex that targets a specific GPCR toward its ultimate effector. Such a complex was proposed for the β2 adrenergic receptor and the L-type Cav1.2 channel17 and between dopamine receptors (D2 and D4) and the inward rectifier Kir3 family and it seems to be relevant to other ion channels as well.18
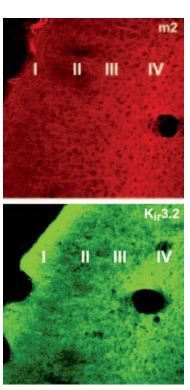
Immunohistochemical distribution of Kir3.2 and M2 in mouse parieto-temporal cortex. Frozen sections (non-consecutive sections) of mouse parieto-temporal cortex were visualized using Anti-GIRK2 (Kir3.2) Antibody (#APC-006) (1:100) and Anti-M2 Muscarinic Receptor Antibody (#AMR-002) (1:100). M2 staining was dense in layer IV, with fibers climbing to layers II-III. Kir3.2 staining was dense in layers IV and I. Overlapping expression of Kir3.2 channel and M2 is seen in cortical layers.
Ion Channels Modulated by GPCRs
Ca2+ Channels
Ca2+ ions regulate many cellular processes such as secretion, proliferation, and apoptosis and are cofactors of many proteins.19,20 In neurons, Ca2+ entry through Cav channels triggers neurotransmitter release. Fast synaptic transmission in the mammalian central nervous system (CNS) is mediated by several highvoltage-dependent Ca2+ channels.21 Five types of Ca2+ channels are expressed in the CNS: the L-type (Cav1), N-type (Cav2.2), P/Q-type (Cav2.1), R-type (Cav2.3), and the T-type (Cav3).16 Each Cav channel is a multimeric protein composed of a pore forming α1 subunit and the auxiliary β (CaVβ), α2δ and γ subunits. There are four known CaVβ subunits, in addition to four α2δ subunits and eight γ subunits. The best characterized Ca2+channels that are regulated by GPCRs are the Ntype and the P/Q-type which have significant roles in neuronal communication.22 This mechanism is the basis of synaptic modulation caused both by endogenous hormones as well as exogenously applied agents (such as analgesia caused by morphine). The identification of the types of Ca2+ channels that are modulated by GPCRs was enabled by the use of specific toxins: ω-Conotoxin GVIA for the N-type channels and ω-Agatoxin-IVA for the P/Q-type channels.23
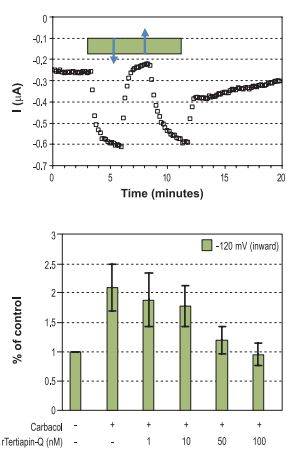
Top: Time course of Kir3 inward current amplitude at -120 mV upon charbacol activation, recorded from Xenopus oocytes heterologously expressing the Kir3.1 and Kir3.4 subunits and the muscarinic M2 receptor. The bar represents period of charbacol (20 μM) perfusion, which activates the channel. Addition of 100 nM Tertiapin-Q(#STT-170) (↓) and wash (↑). Bottom: Summary of the effect of 20 μM charbacol and the dose dependency of the charbacol activated inward currents (at -120 mV) to inhibition by Tertiapin-Q.
N-type and P/Q-type Ca2+ channels are negatively regulated by many different GPCRs by the two distinct mechanisms mentioned above. The indirect mechanism is voltage-independent and activates multiple intracellular signaling pathways, most of them involving second messenger and channel phosphorylation. Cell type, location of the channel within the neuron, and the type of neurotransmitter determines which G-protein subunit, Gαq, Gαi and/or Gβγ mediates the response.13 The second mechanism is a voltage dependent pathway. It inhibits the N-type and P/Q-type Ca2+ channels (but not the L-type) and involves direct interaction of the Gβγ with the α1 subunit. The degree of inhibition is dependent on both the combination of the Gβγ subunits and the type of channel.24
A binding site for Gβγ that was demonstrated to be crucial for the voltage dependent inhibitory mechanism was found in the I-II linker domain (L1). The Gβγ binding site is adjacent to and slightly overlaps the binding site for the Cavβ of the channels and might give some clue as to the mechanism by which Gβγ exerts its inhibitory effect.13,22,25 However, additional binding sites for the Gβγ subunit were found:
1. At the C-terminus of the α1 subunit, another Gβγbinding site that also overlaps with a Cavβ binding site, has been identified.13,22,25
2. At the N-terminus where a putative binding sequence for the Gβγwas identified and was found to be critical for Cav2 voltage dependent inhibition.13,26,27
Although three potential binding sites were described (Fig. 2), indirect kinetic studies pointed to only a single Gβγ binding site per channel raising the possibility that a combination of different Gβγ binding segments on the α1 subunit create the actual binding site for the Gβγ.13
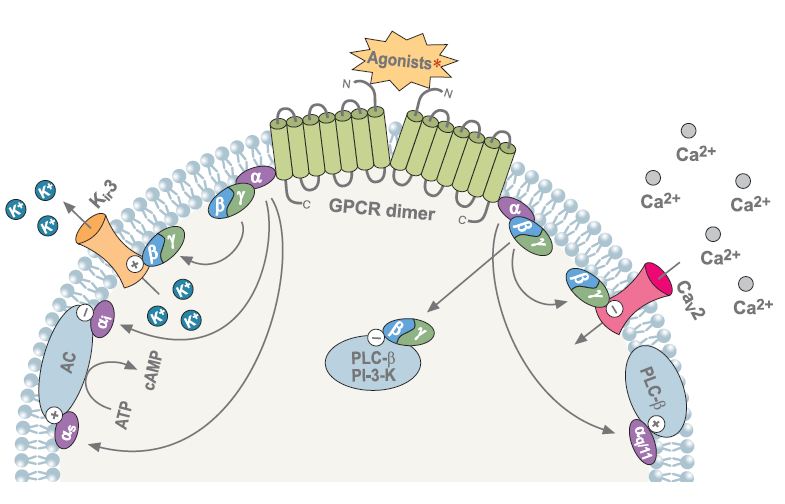
Following agonist stimulation of GPCRs, the Gα and Gβγ subunits dissassociate and either subunit can modulate a diverse array of effectors. “+” indicates activation, “-” indicates inhibition.
∗Agonists: somatostatin, cannabinoid, adrenalin, acetylcholine, GABA, dopamine, serotonin and others.Abbreviations: PLC-β- phospholipase C-β, PI-3K-phosphoinositide-3-kinase, AC- adenylyl cyclase.
K+ Channels
There are four known K+ channel families:
1. The voltage dependent K+channels designated as Kvchannels, which consist of twelve subfamilies.
2. The two pore domain channels, the K2P, which consist of fourteen subfamilies.
3. The calcium activated K+channels, KCachannels, which consist of five subfamilies.
4. The inward rectifier K+channels, the Kir, which include seven subfamilies, designated Kir1 – Kir7 with fifteen members.
GPCRs modulate a number of K+ channels. However, the most intensively studied and characterized are the K+ inward rectifier Kir3 subfamily (Kir3.1- Kir3.4). A low basal level of activity, which is dramatically increased following stimulation of Gi/o-coupled receptors characterizes the Kir3 channels.28,29
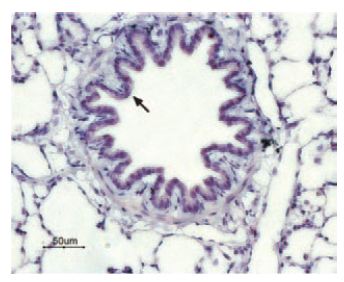
Immunohistochemical staining of P2Y13 receptor in rat lung. Paraffin embedded sections of rat lung were visualized with Anti-P2Y13 Receptor Antibody (#APR-017) (1:50). Strong and highly specific staining is shown in bronchiolar epithelium cells (black arrow). Universal Immuno-alkaline-phosphathase Polymer followed by New Fuchsin Substrate (Histofine, Nichirei corp) was used for the color reaction. Counterstain is Hematoxilin.
Inward rectifier K+ channels (Kir3, formerly named the GIRK channels: G-protein Inward Rectifier K+ channels), play an important role in maintaining the resting membrane potential by regulating the action potential duration and membrane excitability.30 A functional Kir3 channel consists of four subunits, which can be either identical, forming a homotetrameric channel or different, forming a heterotetrameric channel. The latter is the most abundant form of functional channel throughout the body.31 Different combinations of Kir3 subunits have been demonstrated in different tissues. The cardiac channels are composed of Kir3.1 and Kir3.4 and are involved in regulation of cardiac rate. Neuronal channels are mainly composed of heterotetramers of Kir3.1/Kir3.2.28,31,32
The Kir3 channels are activated by various GPCRs that interact selectively with pertussis toxin (PTX)- sensitive Gαi/o proteins.9 They are activated when Gβγ subunits bind directly to the N- and C-terminal regions of each subunit. The channel can also be activated by phosphatidylinositol biphosphate (PIP2) which is essential for the proper gating of the channel following Gβγ activation.33 RGS proteins are responsible for the fast deactivation of the Kir3 channels. The location of the four Gβγ binding sites per Kir3 channel have been suggested to be at both the N- and the C-terminal regions. A synergistic enhancement between these four regions was suggested as a result of the close physical association of these regions.7,34 Among the GPCRs that have been reported to modulate Kir3 channels are the muscarinic receptors, P2Y receptors, somatostatin receptors, dopamine receptors and the GABA(B) receptors.
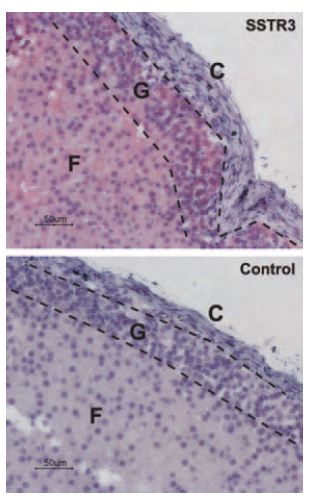
Immunohistochemical staining of somatostatin receptor 3 (SSTR3) in rat adrenal cortex. Paraffin embedded sections of rat adrenal gland were visualized using Anti-Somatostatin Receptor Type 3 Antibody (#ASR-003) (1:50). Strong staining is evident in both glomerular (G) and fasciculate (F) zones but not in connective tissue of the capsule (C). Universal Immunoalkaline-phosphathase Polymer followed by New Fuchsin Substrate (Histofine, Nichirei Corp) was used for the color reaction. Counterstain is Hematoxilin.
GPCRs Involved in Ca2+ and K+ Channel Modulation
As mentioned previously, a broad spectrum of GPCRs modulates ion channel activity. Adrenaline, glutamate, serotonin, acetylcholine (muscarinic), dopamine, opioids, P2Y, cannabinoid, somatostatin, and the GABA(B) receptors have all been shown to modulate Ca2+ and K+ channel actvity.13-16,35 Several of these will be briefly reviewed below.
The Muscarinic Receptors
Muscarinic receptors are metabotropic acetylcholine receptors, and are widely distributed throughout the human body. In the CNS, there is evidence that muscarinic receptors are involved in motor control, temperature regulation, cardiovascular regulation, learning and memory.36 There are five known muscarinic receptors: m1-m5. All five receptors are expressed in the brain, and each with a different distribution pattern. Muscarinic receptors can modulate both Ca2+ and K+ channels.
The muscarinic signaling pathway appears to be the most complex, and includes a large number of components such as: G-proteins, enzymes, second messengers, accessory proteins, cellular growth factors, transformation factors and ion channels.29 Muscarinic receptors modulate Ca2+ channel inhibition through both mechanisms, the voltage independent and the voltage dependent. Accumulated data from pharmacological experiments demonstrated that in rat, as well as in mouse, the m1 receptor is involved in the voltage independent mechanism of inhibition, depressing several high voltage dependent Ca2+ currents.37 The m1 receptor modulates both Ntype and L-type channels through the voltage independent mechanism involving a cytoplasmic second messenger.37 This mechanism is related to muscarinic receptors m1, m3 and m5.
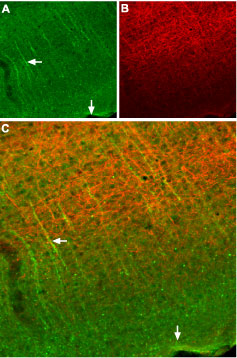
Immunohistochemical staining of somatostatin receptor 2 (SSTR2) in mouse cortex. Frozen sections of mouse cortex were visualized using Anti-Somatostatin Receptor Type 2 (extracellular) Antibody (#ASR-006), (1:100). (A) SSTR2 appears in neural processes (green, horizontal arrow) that are perpendicular to the cortical surface (vertical arrow). (B) Staining of axons with mouse anti-Neurofilament 200 (NF200, red) appears mostly in the deep layers. (C) Confocal merge of SSTR2 and NF200 images suggests that SSTR2 is not present in axons that run in the deep layers but rather in neuronal dendrites that ascend toward upper layers of cortex (vertical arrow).
On the other hand, muscarinic receptors m2 and m4 are considered to mediate Ca2+ channel inhibition through the voltage dependent mechanism, by direct binding of the Gβγ subunit to the α1 subunit of the channel. Although both m2 and m4 are considered to modulate Cav channels by the same mechanism, a recent paper describes an experiment done with knockout mice which revealed that in m4 deficient mice the voltage dependent mechanism was not affected, while m2 knockout mice lacked the voltage dependent pathway.37
Although the general mechanisms described above are well established, in different tissues and cell lines different types of muscarinic receptors participate in the activation of different pathways.
In NIH-3T3 cells, m1 activation did not affect the Cav3 (T-type channel) whereas activation of m3 and m5 receptors enhanced channel activity. m2 receptor affected channel activity only after elevation of cAMP levels by pre-treating the cells with Forskolin, resulting in the inhibition of the channel.
In HEK293 cells, expressing Cav2.3 channels, the R-type channel was shown to be both facilitated and inhibited by m1 and m2 receptors. However, inhibition was much weaker than facilitation for the m1 receptor, while for m2 receptors, inhibition was much more pronounced than activation.29 The differential effects of muscarinic receptors on the R-type channel expressed in several cell lines, indicate that there might be differences in the susceptibility of the channel to modulation by muscarinic receptors. Facilitation and inhibition by the same receptors was probably exerted via different pathways as was indicated by their response to various inhibitors.29 This might be correct with respect to other Ca2+ channels as well.
Muscarinic receptors, as mentioned before, also modulate K+ channels. Stimulation of muscarinic receptor m2 by acetylcholine in the heart results in activation of the Kir3 channels through the membrane delimited mechanism. The Gβγ subunits activates the Kir3.1/Kir3.4 channels by directly binding to their N- and C-termini, causing a slower heartbeat.
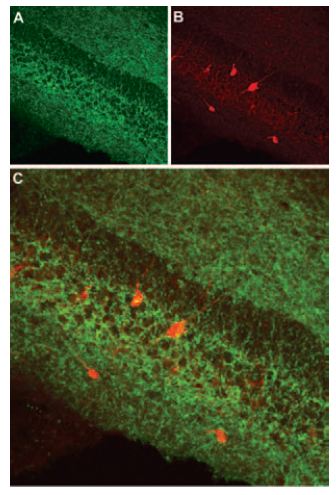
Immunohistochemical staining of cannabinoid receptor 1 (CB1R) in mouse hippocampus. Frozen sections of mouse hippocampus were visualized using Anti-Cannabinoid Receptor 1 (extracellular) Antibody (#ACR-001) (1:100). (A) CB1R appears in the pyramidal and infra-pyramidal layer (green). (B) Staining of interneurons with mouse anti-parvalbumin antibody (PV, red). (C) Confocal merge of CB1R and PV does not demonstrate presence of CB1R in GABAergic cells.
Stimulation of m2 receptor by carbachol, in Xenopus oocytes injected with mRNA of Kir3.1, Kir3.4 and m2, activated Kir3 channels. Activation was reversed by the use of Tertiapin, a specific blocker for Kir channels (which blocks both the Kir3.1 and Kir3.4 channels).38 (Fig. 3,4)
m1, which is a Gq/11 coupled receptor, was shown to inhibit both basal and receptoractivated Kir3 currents.28,29
The exact mechanism is not clear. However, since activation of Kir3 channels occurs through direct binding of the Gβγ to the channel, it was suggested that m1 inhibition occurs either by inactivating the Gi protein or perhaps by directly targeting the Kir3 channel.28
m1 receptor, in sympathetic neurons, was also found to inhibit the Kv7 (KCNQ) family (M-type K+ channels), which might be a result of local depletion in PIP2 which is needed in order to keep the channel in its open state.39
The P2Y Receptors
Extracellular nucleotides such as ATP and UTP are usually co-released with other transmitters from cells and interact with cell surface P2Y receptors to produce a broad range of physiological responses: cardiac function, platelet aggregation and smooth muscle cell proliferation.40
Eight mammalian P2Y receptors are known: the P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13 and P2Y14. Two subfamilies are defined within the P2Y family, which are distinct from each other on the basis of sequence identities and signaling properties:
1. The P2Y1 subfamily includes the P2Y1, P2Y2, P2Y4, P2Y6 and P2Y11 receptors and is coupled to phospholipase C, mediating increases in inositol triphosphate (IP3).
2. The P2Y12 subfamily consists of the P2Y12, P2Y13, and the P2Y14 receptors and is coupled to inhibition of AC.
All P2Y receptor subtypes are expressed in brain tissue and all are capable of modulating ion channels (with the exception of P2Y14 that has not been characterized).
In primary sympathetic neurons, P2Y1, P2Y2, P2Y4 and P2Y6 were all capable of inhibiting both Ca2+ and K+currents. Ca2+ channel (N-type) inhibition is achieved by both mechanisms: the voltage dependent and the voltage independent. Inhibition of K+ channels (Kv7.2, 3 and 5) by those receptors is mediated by PLC and IP3-dependent increases in intracellular Ca2+.41,42 Although P2Y1, P2Y2 and P2Y6 receptors were shown to equally inhibit both Ca2+ and K+ current, coupling of the P2Y4 receptor to KV7 channels was much more efficient than to Ca2+ channels.42 The P2Y12 receptor, in contrast, inhibits only Ca2+ channels through the voltage dependent mechanism.41,43,44 It seems that the P2Y13 receptor, like P2Y12, also inhibits N-type Ca2+channel via the Gβγ subunit possibly, through the voltage dependent mechanism, however, this is not well established.45
The Somatostatin Receptors
Somatostatin is a small cyclic peptide that is widely expressed throughout the CNS and peripheral tissues.46 In peripheral tissues, somatostatin exerts inhibitory effects on secretion processes, whereas in the brain it acts as a neurotransmitter in both a stimulatory and inhibitory manner.45,46 Six receptors, encoded by five genes, underlie the action of somatostatin: SSTR1, SSTR2 (with two known alternative spliced isoforms; SSTR2a, SSTR2b), SSTR3, SSTR4 and SSTR5.46,47 All somatostatin receptors are expressed in the CNS, and in endocrine/exocrine glands.48
Activation of somatostatin receptors is coupled to multiple signaling pathways. All somatostatin receptors inhibit AC activity while at the same time are able to selectively activate other transduction targets.48 SSTRs also modulate several ion channels. Among them are the Kir channels, Kv channels, Ca2+ activated K+ channel (KCa) and the high voltage activated L- and N-type Cav channels.48,49 However, inconsistent results were reported with SSTR1 and SSTR2 in heterologous systems in respect to their coupling to AC and to ion channels. In pituitary cells, somatostatin receptor was found to activate K+ channels and to inhibit voltage dependent Ca2+ channels.50 In the pancreatic β-cell line MIN-6, SSTRs activated two types of inward rectifier K+ channels; the KATP and Kir3 channels.51 Somatostatin receptors were also shown to modulate the transient receptor potential vanilloid 1 channel (TRPV1).52 DRG cells expressing TRPV1 also express, in most cases, SSTR2.
In vivo behavioral studies indicated that SSTRs can modulate the TRPV1 channel and this is further supported by in vitro experiments.52

Immunohistochemical staining of GABA(B) R1 in mouse hippocampus. Frozen sections of mouse hippocampus was visualized using Anti-GABA(B) R1 (extracellular) Antibody (#AGB-001) (1:100). (A) GABA(B) R1 (green) appears in neurons in the CA3 field and in the dentate granule layer (short arrows) and dendrites of CA3 pyramidal neurons (long arrows). (B) Staining with mouse anti GAP43 identifies stratum lacunosum moleculare (SLM). (C) Confocal merge of GABA(B) R1 and PV suggests presence of GABA(B) R1 in pyramidal neurons.
The Cannabinoid Receptors
Cannabinoids have been used in eastern medicine for many years as pain relievers.53 To date, two specific cannabinoid receptors, known as CB1 and CB2, have been identified. Both of them are coupled to the Gi/o G-protein.54,55 CB1 was shown to modulate Ca2+ and K+ ion channels in different tissues and cell lines.54-57Inhibition of pre-synaptic N-type and P/Q-type Ca2+ channels was demonstrated by several cannabinoids (endocannabinoids as well as exogenously applied) in heterologously expressed mammalian neurons,55cultured hippocampal neurons,57,58 neuroblastoma-glioma cells (N-type only) and in exogenously expressed CB1 receptor in pituitary tumor cells (P/Q-type).59 Endocannabinoid stimulation of the CB1 receptor was also shown to activate the Kir3 channels both in pituitary tumor cells and in heterologously expressed mammalian neurons, where it was shown that ion channel modulation by the CB1 receptor is also dependent on expression levels of the receptor.55 It has been suggested that different agonists of CB1 receptor might affect the selectivity of the interaction between the receptor and the G-proteins by inducing different conformational states of receptor.60
The GABA(B) Receptors
GABA(B) receptors mediate slow synaptic inhibition in the brain and spinal cord and are activated by gamma aminobutiric acid (GABA) which is a major inhibitory neurotransmitter.
GABA(B) receptors are coupled to the pertussis toxin-sensitive G-proteins, the Gαi/o.61,62 The functional GABA(B) receptor is a heterodimer, which consist of two subunits, GABA(B) R1 and GABA(B) R2. Cell surface expression is also dependent on the receptor being a heterodimer.61,62 GABA(B) R2 was shown to be essential for trafficking of the receptor to the cell surface as well as for activation of effector systems, while GABA(B) R1 was found to be important for the ligand binding along with GABA(B) R2.63-65 Inhibition of Ca2+ (N-type and P/Q-type) channels on one hand and on the other hand activation of Kir3 channels by GABA(B) receptors has been demonstrated, probably by the same voltage dependent mechanism involving direct binding of the Gβγ subunits.66,67
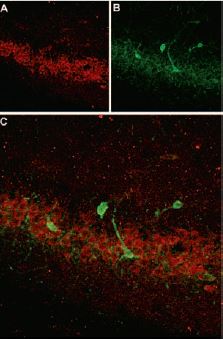
Immunohistochemical staining of somatostatin receptor 4 (SSTR4) in rat hippocampus. Frozen sections of rat hippocampus were visualized with Anti-Somatostatin Receptor Type 4 (extracellular) Antibody (#ASR-004) (1:100). (A) SSTR4 appears in the pyramidal layer (red). (B) staining of interneurons with mouse anti-parvalbumin antibody (PV, green). (C) Confocal merge of SSTR4 and PV demonstrates separate localization in neocortex.
Summary Remarks
Ion channel modulation by GPCRs is much more diverse and complex than what was first expected. The diversity of ion channels along with the large number of GPCRs, and G-protein combinations creates a diverse array of signaling pathways which control cellular functions. The complexity is growing while new functions are constantly being discovered, unmasking known orphan GPCRs. Both ion channels and GPCRs have become targets in the search for therapeutic drugs in cancer, heart diseases, obesity among others.
References
- Fredriksson, R. et al. (2003) Mol. Pharmacol. 63, 1256.
- Alberts, B. et al. (2002) The Cell, Fourth Edition. p.831, 852.
- Kroeze, W.K. et al. (2003) J. Cell. Sci. 116, 4867.
- Wess, J. (1997) FASEB. J. 11, 346.
- Hur, E.M. and Kim, K.T. (2002) Cell. Signal. 14, 397.
- Neves, S.R. et al. (2002) Science 296, 1636.
- Sadja, R. et al. (2003) Neuron 39, 9.
- Riddle, E.L. et al. (2005) Circ. Res. 96, 401.
- Zhang, Q. et al. (2002) J. Physiol. 545, 355.
- Capener, C.E. et al. (2002) Human Mol. Gen. 11, 2425.
- Hubner, C.A. and Jentsch, T.J. (2002) Human. Mol. Gen. 11, 2435.
- Green, W.N. and Millar, N.S. (1995) Trends Neurosci. 18, 280.
- Dascal, N. (2001) Trends Endocrinol. Metab. 12, 391.
- Magoski, N.S. and Kaczmarek, L.K. (1998) J. Physiol. 509, 1.
- Diverse-Pierluissi, M. (2005) Science’s STKE. 2005, tr21.
- Mark, M.D. and Herlitze, S. (2000) Eur. J. Biochem. 267, 5830.
- Davare, M.A. et al. (2001) Science 293, 98.
- Lavine, N. et al. (2002) J. Biol. Chem. 48, 46010.
- Bonilla, M. and Cunningham, K.M. (2002) Sci. STKE 127, PE17.
- Wu, X. et al. (2004) J. Biol. Chem. 279, 43392.
- Iwasaki, S. et al. (2000) J. Neurosci. 20, 59.
- Dolphin, A.C. (2003) Pharmacol. Rev. 55, 607.
- Elmslie, K.S. (2003) J. Bioenerg. Biomemb. 35, 477.
- Zhou, J.Y. et al. (2000) J. Neurosci. 20, 7143.
- Cooper, C.B. et al. (2000) J. Biol. Chem. 275, 40777.
- Page, K.M. et al. (1998) J. Neurosci. 18, 4815.
- Canti, C. et al. (1999) J. Neurosci. 19, 6855.
- Hill, J.J. and Peralta, E.G. (2001) J. Biol. Chem. 276, 5505.
- Lanzafame, A.A. et al. (2003) Receptors Channels 9, 241.
- Costa, A.C.S. et al. (2005) J. Neurosci. 25, 7801.
- Ivanina, T. et al. (2003) J. Biol. Chem. 278, 29174.
- Kurachi, Y. and Ishii, M. (2004) J. Physiol. 554, 285.
- Rishal, I. et al. (2005) J. Biol. Chem. 280, 16685.
- Stanfield, P.R. et al. (2002) Rev. Physiol. Biochem. Pharmacol. 145, 47.
- Brown, A.M. (1991) FASEB. J. 5, 2175.
- Caulfield, M.P. et al. (1994) J.Physiol. 477, 415.
- Shapiro, M.S. et al. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 10899.
- Jin, W. and Lu, Z. (1998) Biochemistry 37, 13291.
- Winks, J.S. et al. (2005) J. Neurosci. 25, 3400.
- Wang, L. et al. (2002) J. Cardiovas. Pharmacol. 40, 841.
- Filippov, A.K. et al. (1998) J. Neurosci. 18, 5170.
- Filippov, A.K. et al. (2003) Br. J. Pharmacol. 138, 400.
- Boehm, S. (2003) Br. J. Pharmacol. 138, 1.
- Lechner, S.G. and Boehm, S. (2004) Purinergic Signaling 1, 31.
- Wirkner, K. et al. (2004) Br. J. Pharmacol. 141, 141.
- Hofland, L.J. and Lamberts, S.W. (2001) Ann. of Oncol. 12, S31.
- Fombonne, J. et al. (2003) Reprod. Biol. Endocrinol. 1, 19.
- Krantic, S. et al. (2004) Eur. J. Endocrinol. 151, 643.
- Akopian, A. et al. (2000) J. Neurosci. 20, 929.
- Chen, L. et al. (1997) J. Biol. Chem. 272, 18666.
- Smith, P.A. et al. (2001) J. Physiol. 532, 127.
- Carlton, S.M. et al. (2004) Pain 110, 616.
- Brooks, J.W. and Farquhar-Smith, W.P. (2003) Br. J. Anaesth. 3, 175.
- Howlett, A.C. (2002) Prostaglandins Other Lipid Mediat. 68-69, 619.
- Guo, J. and Ikeda, S.R. (2004) Mol. Pharmacol. 65, 665.
- Howlett, A.C. et al. (2002) Pharmacol. Rev. 54, 161.
- Twitchell, W. (1997) J. Neurophysiol. 78, 43.
- Sullivan, J.M. (1999) J. Neurophysiol. 82, 1286.
- Mackie, K. et al. (1995) J. Neurosci. 15, 6552.
- Glass, M. and Northup, J.K. (1999) Mol. Pharmacol. 56, 1362.
- Margeta-Mitrovic M. et al. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14649.
- Margeta-Mitrovic M. et al. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14643.
- Couve, A. et al. (1998) J. Biol. Chem. 273, 26361.
- Fillppov, A.K. et al. (2000) J. Neurosci. 20, 2867.
- Robbins, M.J. et al. (2001) J. Neurosci. 21, 8043.
- Wang, X. and Lambert, N.A. (2000) J. Neurophysiol. 83, 1073.
- Wetherington, J.P. and Lambert, N.A. (2002) J. Physiol. 544, 459.