Glial-Derived Neurotrophic Factor (GDNF) is the major member of the GDNF family protein which is distantly related to the TGF-β superfamily. GDNF elicits its cellular and physiological effects by binding RET, a tyrosine receptor kinase and GPI-linked co-receptors, belonging to the GDNF receptor α family. GDNF is a disulfide-bonded neurotrophic factor that is produced as an inactive precursor. The presignal peptide is cleaved upon protein secretion, followed by the cleavage of the pro-signal probably at the target’s cell surface to produce functional mature homodimers. Here, we briefly describe some activities and downstream signaling of GDNF and the contributions of Alomone Labs in this field.
Introduction
Since the discovery of GDNF in 199313, numerous in vitro and in vivo studies identified it as a survival factor, and a factor required for axonal growth for midbrain dopaminergic neurons. GDNF was also found to be important in maintaining several neuronal populations in the central nervous system, including hippocampal regions1 and motoneurons11. GDNF is also involved in regulating the differentiation of many peripheral neurons, including sympathetic, parasympathetic, sensory and enteric neurons13,10,2,12,17. Recombinant human GDNF protein (#G-240) is a particularly potent factor for the induction of tyrosine hydroxylase expression which results in the increase of catecholamine synthesis18,6. GDNF was shown to enhance the reuptake of dopamine in the dopaminergic neuron in the midbrain and in the nigrostriatal pathway of the brain, those neurons that degenerate in Parkinson’s disease. Indeed, GDNF raised great expectations as a potential therapeutic agent for the treatment of neurodegenerative diseases, including Parkinson’s disease13. A recent trial indicated that GDNF, directly infused to the forebrain Putamen prevents neuronal loss, reduces brain damage and improves some motor deficits in several animal models of Parkinson’s disease including primates and human, and does not cause any significant side-effects13,10,2,16. Moreover, GDNF could enhance synaptic efficacy of dopaminergic but not GABAergic neurons in culture3 (Figure 1). In the brain, GDNF was demonstrated to act as a rescuing factor thereby reducing apoptosis, whereas BDNF accelerated aluminium induced apoptosis19. Further critical roles of GDNF have been found outside the nervous system in the regulation of kidney morphogenesis and spermatogenesis16.
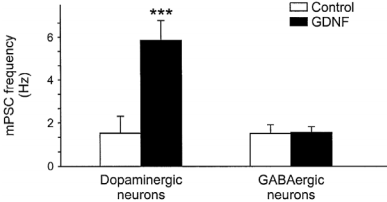
Histogram of the mean miniature synaptic event frequency. The mean frequency was larger after Recombinant human GDNF protein (#G-240) treatment in dopaminergic neurons (n=18) the same treatment in GABAergic neurons did not change the mean miniature synaptic event frequency.
Adapted from reference 3 with permission of Blackwell Publishing Ltd.
GDNF Signaling Pathways
GDNF and its family members signal via the receptor tyrosine kinase RET in combination with one of several GPI-linked co-receptors belonging to the GDNF Receptor α family (GDNFRα, GFRα, or RETL) to create a cell-surface complex. Activation of the complex by GDNF leads to RET autophosphorylation7 (see Figure 4) and cell differentiation in PC12 cells expressing the receptor8. GDNF-activated RET could also induce different biological responses such as morphological transformation, proliferation, cell migration, neurite elongation and neurite branching. In cells lacking the RET receptor, alternative mechanisms which involve neural cell adhesion molecule (NCAM) was suggested for GDNF activities8,16. In the hippocampus, GDNF was shown to act at the CA3 region through the activation of RET-ERK pathway without involving the NCAM-mediated pathway1 (Figure 2). In this region, GDNF has a fundamental role in protecting cells from NMDA induced neurodegeneration to a greater extent than in the hippocampal CA1 region. Confocal images revealed that upon NMDA exposure, RET-ERK activation occurs in microglial cells in both regions following GDNF exposure1 (Figure 3).The tyrosine kinase activity of RET stimulated by GDNF was abolished in the presence of a specific RET inhibitor5. RET signaling was enhanced when RET was co-expressed with the adaptor protein SH2B1β. In fact, co expression of both proteins potentiated GDNF differentiating and transforming abilities and protected GDNF-stimulated RET from specific RET inhibitors1.
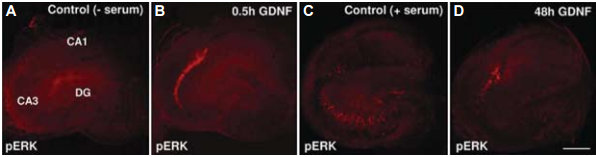
Distribution of pERK immunoreactivity in control serum-free-exposed Organotypic Hippocampal Slice Cultures (OHSCs) for 30 minutes (A), in control serum exposed OHSCs (C), in mature OHSCs exposed to acute (B) or to chronic Recombinant human GDNF protein (#G-240) exposure (D).
Adapted from Boscia, F. et al. (2009) PLoS One 4, 6486.
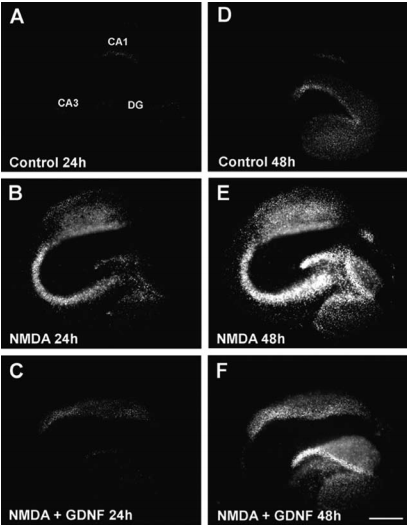
Propidium Iodide (PI) fluorescence staining patterns observed in representative OHSCs 24 h (A-C) and 48h (D-F) following their exposure to control condition (A,D), NMDA (B,E) or NMDA + Recombinant human GDNF protein (#G-240) (C,F).
Adapted from Boscia, F. et al. (2009) PLoS One 4, 6486.
The RET receptor gene is alternatively spliced to yield two main isoforms, RET9 and RET51, which differ in their carboxyl terminus but not in their biochemical activity. Mutation at Tyr1062 abrogated the transforming activity caused by GDNF in cells expressing both isoforms7. This mutation, however, did not abolish activation and cell scattering and branching morphogenesis in RET51 expressing cells, whereas impaired these biological effects in RET9 expressing cells7. Additionally, GDNF-induced biological effects of RET51 were inhibited by the simultaneous abolition of Tyr1062 and Tyr1096 (which are both docking sites for downstream signaling). However, Tyr1096 could substitute for the biochemical functions of Tyr1062 in response to GDNF7 as demonstrated in Figure 4. As Tyr1096 is the only known docking site for GRB2, this data suggests that Tyr1096 is necessary for GDNF-mediated branching, through the binding of GRB2 protein7.
Intracellular Ca2+ fluctuations by GDNF play an important role in the activation of some members of the signaling pathways related to neuronal survival such as ERK and Akt7 (Figures 4 and 5). Stimulation of spinal cord motoneuron cells with GDNF led to moderate intracellular Ca2+ mobilization (Figure 5) induced by calmodulin activation14. Calmodulin inhibition reduced PKB phosphorylation and prevented survival of motoneuron cells induced by GDNF and eventually led to apoptosis14. However, GDNF-mediated survival of sympathetic neurons was completely prevented in RET knock in mice mutated in Tyr1096, while no effect on cell survival was observed in RET knock in mice mutated in Tyr981, which exhibited abrogation of PKB phosphorylation. In contrast, silencing of B-Raf abolished cell survival suggesting that the B-RafIKK pathway is responsible for the GDNF survival effect in sympathetic neurons, independently from the PI3K-ERK pathway9.
Carcinomas are often associated with oncogenic activation. Papillary thyroid carcinoma cells highly express oncogenic derivatives of RET receptor as well as its isoforms, and its associated GFRα2 co-receptor13,10. In these cells, GDNF treatment induced RET tyrosine phosphorylation, its subsequent signal transduction and cell proliferation indicating that RET and RET mutants could be active in thyroid follicular cells4,5. In contrast, in human neoplastic chromaffin cells GDNF-activated RET was differentiative rather than mitogenic15.
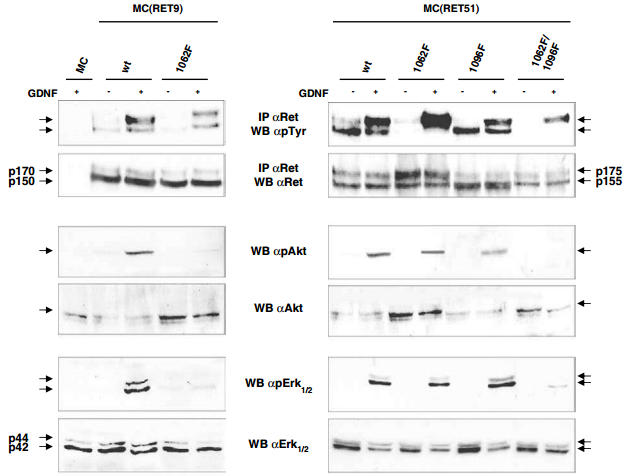
Western blot analysis of the Recombinant human GDNF protein (#G-240) -activated signaling pathways in neuroepithelioma-MC cells expressing RET isoforms. Parental and MC clones stably expressing wild type or mutated RET proteins were untreated or treated with 100 ng/ml hGDNF for 10 min. Cell lysates were immunoprecipitated with anti-RET and blotted with anti-pTyr or anti-RET. For ERK1/2 and Akt, phosphorylated activated forms were detected with anti-phospho ERK1/2 and Akt and anti-ERK1/2 and Akt antibodies on total cell lysates.
Adapted from reference 7 with permission of Macmillan Publishers Ltd.
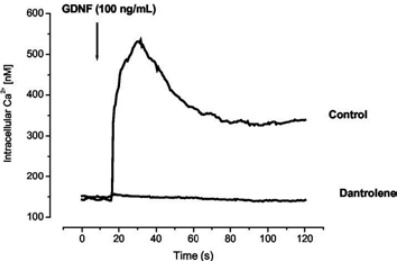
Fluo-4 loded MTNs were stimulated with 100 nM Recombinant human GDNF protein (#G‑240) and intracellular level of Ca2+ was measured. The figure presents traces of intracellular levels of Ca2+ in the presence or absence of the store operated Ca2+ inhibitor – dantrolene (40 μM).
Adapted from reference 14 with permission of The American Society for Biochemistry and Molecular Biology.
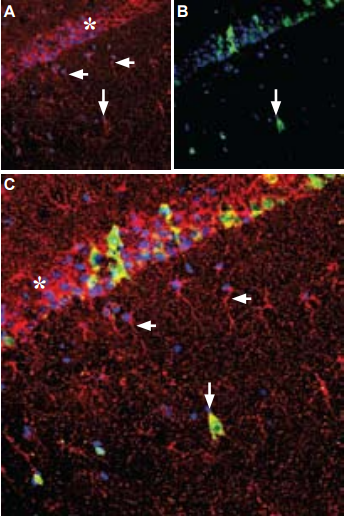
A. Immunohistochemical staining of rat hippocampus frozen sections with Anti-GDNF Antibody (#ANT-014), (1:100), (red) is apparent in the pyramidal layer (asterisk), in astrocytic fibers (horizontal arrows) and in some interneurons (vertical arrow). B. Calbindin D28k (green) appears in a subset of cells in the pyramidal layer. C. Merge of GDNF and calbindin demonstrates some co-localization in neurons. DAPI is used as the counterstain (blue).
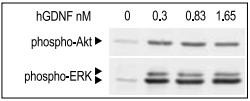
SHSY-5Y cells were serum starved for 2 h, and then stimulated with different concentrations of Recombinant human GDNF protein (#G-240) for 10 min. The cell proteins were resolved by SDS PAGE and detected with phospho-(Ser 473)- Akt and phospho-ERK1/2 antibodies.
References
- Boscia, F. et al. (2009) PLoS One 4, e6486.
- Brundin, P. (2002) Brain 125, 2149.
- Bourque, M.J. et al. (2000) Euro. J. Neurosci. 12, 3172.
- Bunone, G. et al. (2000) Cancer Res. 60, 2845.
- Carlomagno, F. et al. (2006) J. Natl. Cancer Ins. 98, 326.
- Copray, J.C. et al. (1999) Dev. Brain Res. 116, 217.
- Degl’Innocenti, D. et al. (2004) Oncogene 23, 7297.
- Donatello, S. et al. (2007) Oncogene 26, 6546.
- Encinas, M. et al. (2008) Cell Death Differ. 15, 1510.
- Gash, D.M. et al. (1996) Nature 380, 252.
- Junger, H. and Varon, S. (1997) Brain Res. 762, 56.
- Koeberle, P.D. and Ball, A.K. (2002) Neuroscience 110, 555.
- Lin, L.F. et al. (1993) Science 260, 1130.
- Pérez-García, J.M. et al. (2004) J. Biol. Chem. 279, 6132.
- Powers, J.F. et al. (1998) Endocr. Pathol. 9, 325.
- Sariola, H. and Saarma, M. (2003) J. Cell Sci. 116, 3855.
- Schäfer, K.H. et al. (1999) J. Interferon Cytokine Res. 19, 527.
- Tanaka, M. et al. (2003) Brain Res. Protoc. 11, 119.
- Yang, S.J. et al. (2004) Mol. Brain Res. 127, 146.