Neurotrophins, like many growth factors, are expressed as precursor proteins which are then truncated and further modified to yield the mature form. The presence of highly conserved sequences in the prodomain of different neurotrophins made it clear that the prodomain must have some biological significance and is not solely present as a protein folding chaperone. This reports aims at briefly summarizing the advances made in the understanding of the cellular functions of proNGF and proBDNF and mostly how Alomone Labs’ extensive neurotrophin product line has assisted and contributed to the advances made.
proNGF and proBDNF Processing
The proNGF precursor is synthesized as 25 and 32 kDa isoforms which are glycosylated to form a major 40 kDa species, while proBDNF is expressed as a ~30 kDa protein.
At the beginning of the century, it was recognized that both proNGF and proBDNF may have a biological role in the central nervous system (CNS)4. Therefore the question regarding the processing mechanisms of proNGF to mature NGF and that of proBDNF to BDNF was raised.
For a long time it was believed that NGF was secreted from cells in its mature form. At a point where proNGF was gaining more attention and more research was being initiated, an elegant study was published where the exact processing of proNGF in mature CNS neurons was worked out4. Using small slices of the frontal and parietal cerebral cortex of young rats, the study showed that NGF is released in the extracellular space in its precursor form in an activity dependent fashion (by stimulating the cortex tissue with either carbachol, glutamate, or KCl). The study further showed using Anti-proNGF Antibody (#ANT-005) that proNGF is secreted from cells along with the processing enzymes and their regulators (Figure 1) in order to generate mature NGF (13 kDa) or degrade it. It was also shown that proNGF secretion is dependent on intracellular Ca2+ stores4.
proBDNF on the other hand, seems to exhibit a much more controversial processing event. Evidence that BDNF was secreted from cells in its precursor form was published by Lee et al.12. Pang et al.16 further reinforced later on by that BDNF is secreted with the prodomain, and once in the extracellular space processed to its mature form. The secretion of proBDNF was yet again detected in mouse hippocampus and was secreted from cultured hippocampal neurons29. In a different study, proBDNF secretion was taken one step further. It was observed that when cultured hippocampal neurons are exposed to high frequency, proBDNF is the prevailing form secreted, as opposed to low frequency exposure which promotes the secretion of the proneurotrophin and its proteolysis to the mature form14. Quite recently, another important paper published contradicting data from that mentioned above, that BDNF is secreted in its mature form from CNS neurons13. It was suggested that these conflicting data may indicate that BDNF/proBDNF secretion depends on the incorporation (or lack of) of glia cells and plasmin inhibitors (a component in the processing of the proneurotrophin to the mature form)14,29.

Localization of tPA (green) and proNGF (red) using Anti-proNGF Antibody (#ANT-005) in cortical pyramidal neurons; colocalization illustrated with merged images (yellow). Scale bar, 20 μm. Adapted from reference 4 with permission of The National Academy of Sciences of the USA (Copyright 2006)
Neurotoxic vs Neurotrophic Effects of proNGF and proBDNF
For a long time, it was believed that the sole purposes of the prodomain of the proneurotrophins are to assist in protein folding and regulate protein secretion17,21.
Mature NGF and BDNF (and other neurotrophins) regulate neurite outgrowth and neuronal survival during development. Both neurotrophins do so by binding two types of cell surface receptors; Trk, tyrosine kinase receptor and p75 neurotrophin receptor (p75NTR), a member of the tumor necrosis receptor family. Most of the signaling and cellular functions of NGF and BDNF can be found in this issue of NGF (see Dovrat Brass and Alon Meir, respectively). It is worth mentioning however that, while NGF and BDNF are known to preferentially bind TrkA and TrkB receptors respectively, they can also bind, albeit with a lesser affinity, to p75NTR. The coexpression of p75NTR with the appropriate Trk receptor increases the neurotrophin-Trk interactions by enabling the discrimination of the different ligands of the Trk receptor family.
The expression of a mutated form of proNGF that is resistant to cleavage, showed for the first time that proNGF may have a biological role since it displayed a fivefold greater affinity to p75NTR than did mature NGF, and showed negligible binding to TrkA12. proNGF also induced apoptosis in smooth muscle cells expressing p75NTR and sympathetic neurons12 and it was demonstrated later on that proNGF signaling via p75NTR is dependent on sortilin15. Using a different mutant of proNGF, also resistant to cleavage, it was shown, contrary to Lee et al.12, that proNGF could bind TrkA and promote neurite outgrowth, albeit with a much lower efficacy when compared to mature NGF7. These two different studies initiated what remains to date controversial aspects regarding the neurotoxic and neurotrophic properties of proNGF.
As work on proNGF picked up pace, it was found that proNGF is the predominant form in the brain and is increased in brains from Alzheimer’s disease (AD) patients6. Also related to proNGF and AD, a study aimed to investigate the roles of proNGF in neuronal viability, neurite outgrowth and Aβ plaque metabolism in AD pathogenesis27. Applying mainly in vitro studies, it was shown that treating cells with Recombinant human proNGF (cleavage resistant) protein (#N-285), a cleavage resistant form of proNGF, promoted cell death, and decreased neurite growth in SH-SYS5 cell line as well as promoted neurite collapse in primary neurons of mice27 (Figure 2). In the same line of work, a recent study was published1 where the neurotrophic versus neurotoxic activities of proNGF may well be put to rest. In this study the authors show that proNGF induces cell death in subpopulations of basal forebrain and peripheral sympathetic neurons of old, but not of young adult rodents. The neurotoxic effects of proNGF in ageing basal forebrain neurons led the authors to monitor the levels of proNGF in the ageing brain as opposed to that of young brains. Using Anti-proNGF antibody, proNGF levels were first examined in human hippocampus homogenate from two different adults which showed that proNGF recognition could be blocked using the negative control antigen and importantly, Alomone Labs Anti-proNGF antibody did not cross-react with neither proNT3 nor proBDNF1 (Figure 3). proNGF levels were measured in the cortex and hippocampus of young adult and aged mice. While no significant differences in proNGF levels were observed in the cortex of young and aged mice, a marked increase in proNGF protein was detected in the hippocampus of aged mice compared to young mice and even more so in aged rats. Levels of sortilin but not of p75NTR were also elevated in aged rats suggesting that both proNGF and sortilin may indeed play an important role in neurodegeneration in old-aged brains1 .
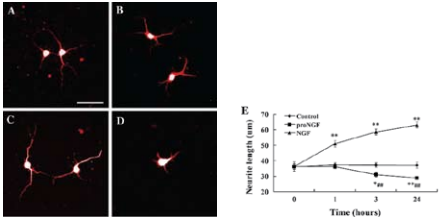
A)-D) Are representative images of neurons stained for MAP2. A) Control neurons before the addition of factors. B) Neurons cultured in the medium for 24 hours without the addition of factors. C) Neurons cultured in the presence of NGF (2 nM) for 24 hours. D) Neurons cultured in the presence of Recombinant human proNGF (cleavage resistant) protein (#N-285), (2 nM) for 24 hours. E) The response of neurites to proNGF, NGF and culture medium in cortical neurons. Images of live neurons were captured at time points 0, 1, 3 and 24 hours following the addition of the factors. The neurite length of neurons was measured with ImagJ software.**P < 0.01 compared to time point 0. Scale bar, 50 μm.Adapted from reference 27 with permission of Springer Science + Business Media
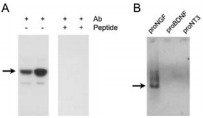
A) Human hippocampus homogenate from two different adults was challenged with Anti-proNGF Antibody (#ANT-005). The panel on the right shows that the signal is completely eradicated when the antibody is preincubated with the antigenic peptide. B) Western blot analysis showing that Anti-proNGF specifically recognizes proNGF. Twenty five nanograms of human recombinant proNGF, Recombinant human proBDNF protein (#B-257), and proNT3 were loaded on different lanes, and the blot was reacted with Anti-proNGF. Arrows represent the 40 kDa proNGF protein.
Adapted from reference 1 with permission of Blackwell Publishing Ltd.
Spinal cord injury (SCI) causes permanent neurological disability by inducing massive cell death. Following a SCI event the increased levels of proNGF play a pivotal role in the apoptosis of oligodentrocytes in a p75NTR-dependent manner2. Using in part Anti-proNGF, an attempt was made in order to understand the cell types producing proNGF and the signaling pathways involved in the production of proNGF following SCI30 (Figures 4 and 5). The authors first showed that proNGF protein levels peak at 5 days following SCI (Figure 4A). Minocycline, a known inhibitor of apoptosis, inhibits the expression of proNGF following SCI as shown in western blot and immunocytochemical analyses (Figures 4B, C). Examining the cell types responsible for proNGF expression five days following SCI, proNGF-positive microglia were found throughout the length of the spinal cord in both the white and gray matters (Figures 4D, E), and only neurons (Figure 4F) and astrocytes (Figure 4G) located near the lesion area. Shamoperated and the negative control showed no expression of proNGF. The authors go on to show that following injury, proNGF is colocalized with activated p38 MAPK (p-p38) in microglia in immunocytochemical studies (Figure 5) and that inhibition of p38 inhibits the p38-dependent expression of proNGF30.
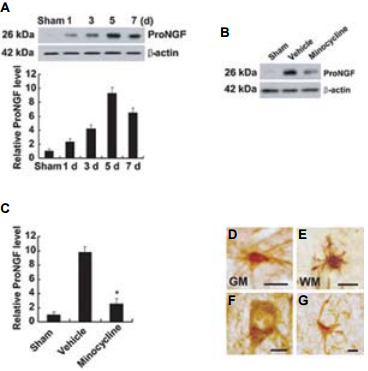
A) Detection of proNGF following 1, 3, 5 and 7 days following SCI using Anti-proNGF Antibody (#ANT-005). B) Minocycline treatment decreased the level of proNGF expression compared with that observed in vehicle-treated control at day 5 after injury. C) Quantitative analysis of western blots shows that minocycline significantly inhibited proNGF expression when compared with that in vehicle control at day 5 post-injury. Values are mean ± SD of three separate experiments. *P < 0.001. D)-G) Immunocytochemical analysis showing that microglia in the white (WM) (D) and gray (GM) (E) matters, and neuron (F) and astrocyte (G) located near the lesion site express proNGF at day 5 after injury. Scale bar, 10 μm.Adapted from reference 30 with permission of The Society for Neuroscience
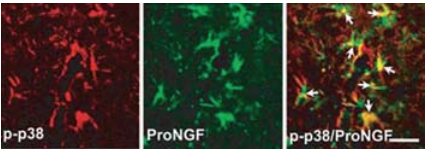
Double-labeled immunocytochemical analysis showing that p-p38 MAPK-positive microglia express proNGF detected with Anti-proNGF Antibody (#ANT-005), (arrows) at 5 days after injury. Scale bar, 20 μm.Adapted from reference 30 with permission of The Society for Neuroscience
Like proNGF, proBDNF was thought to have an insignificant function in cells, until findings showed that proBDNF induces apoptosis in peripheral neurons23, and helps with long-term depression as opposed to long-term potentiation induced by mature BDNF in the hippocampus28.
Hearing loss is a common disability and is caused mostly by old age or by ototoxic drugs such as aminoglycoside antibiotics9. Sensory hair and supporting cells in the organ of Corti, which are affected upon hearing loss, provide neurotrophic support to primary auditory neurons (spiral ganglion). These neurons express both Trk and p75NTR. Indeed, mutant mice with TrkB and TrkC deletions suffer from significant loss of spiral ganglion neurons (SGN) as well as innervation defects in the cochlea during development20. As both sets of receptors are expressed in SGNs, a study was done in order to establish to what extent each of these receptors is involved with neuronal death in degenerating SGNs in an in vivo system22. Using aminoglycoside antibiotics, to induce hearing loss in rats, the expression of p75NTR was found to be up-regulated using Mouse Anti-Rat p75 NGF Receptor (extracellular) Antibody (#AN-170) and that of TrkB was down-regulated. Along with these results, proBDNF was also found to be up-regulated using Anti-proBDNF Antibody (#ANT-006) in agreement with the observed increase in p75NTR 22. However, along with the bona fide proBDNF, an additional band was detected in western blot analysis, termed as a truncated form of proBDNF. The study raised the possibility that perhaps the increase in proBDNF expression may lead to the secondary SGN degeneration observed repeatedly22.
Sortilin has recently been identified as the protein responsible for the apoptosis inducing effect of both proNGF and proBDNF through p75NTR 15,23. To selectively address the contribution of sortilin and proneurotrophins to neuronal viability, a sortilin deficient mouse (Sort-/-) was generated11. SCG neurons from wild-type mice and sortilin deficient mice were exposed to Recombinant mouse proNGF protein (#N-250) as well as Recombinant mouse proBDNF protein (#B-240) and their ability to induce apoptosis was compared. SCG neurons from wild-type mice were protected from apoptosis when NGF was applied but not when the cells were incubated with proNGF or proBDNF (Figure 6). While p75NTR expression was not altered in Sort-/- cells, application of proNGF and proBDNF did not induce an apoptotic response, indicating that sortilin is in fact essential for the killing activity of proNGF and proBDNF observed in wild-type cells. Neurons deficient in p75NTR displayed similar effects. This study showed genetic proof for a role of proNGF and proBDNF complexed with sortilin and p75NTR in the regulation of neuronal viability in vivo11.
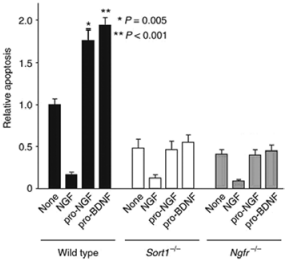
Proneurotrophin-induced apoptosis in SCG cultures. Cells were cultured for 5 days in the absence or presence of NGF (10 ng/ml), Recombinant mouse proNGF protein (#N-250), (5 ng/ml) or Recombinant mouse proBDNF protein (#B-240), (5 ng/ml). After 36 hours, apoptotic cells were scored by TUNEL staining. The total number of cells counted per condition varied between 400 and 600. Apoptosis in wild-type cells in the absence of any addition amounted to ~20% of the counted cells. Shown are the means ± SEM (n = 6). * P = 0.005, ** P < 0.001.Adapted from reference 11 with permission of Macmillan Publishers Ltd.
The induction of prolonged epileptic seizures by pilocarpine introduction results in increased cellular levels of BDNF and both necrotic and apoptotic death18,19,10,5. In a recent study, induced epileptic seizures were accompanied by a significant decrease in TrkB, with an increase in p75NTR expression (probably a reason for which BDNF could also induce cellular death)24. The p75NTR receptor could be immunoprecipitated with proBDNF (for which levels also increased post-seizure) using Anti-proBDNF Antibody. Using the same experimental system the same authors also showed that in vivo administration of Anti-proBDNF Antibody in animals subjected to status epilepticus for three hours, manifest a neuroprotection effect25 (Figure 7).
Active BDNF is also produced in B cells3. In a recent study, the expression of both BDNF and proBDNF was monitored in these cells in order to evaluate whether their expression is manifested by a cellular activity8. Indeed, in immunocytochemical studies, a strong cytoplasmic staining of proBDNF was detected using Anti-proBDNF. Western blot analysis also detected proBDNF in the culture media8, strongly suggesting that B cells also secrete proBDNF. The p75NTR receptor forms disulfide-bridged dimers at the cell surface, independently of neurotrophin binding through a highly conserved cysteine residue in the transmembrane domain. This disulfide bridge formation is essential for the downstream signaling of p75NTR upon neurotrophin activation. In a recent study, the possibility of activating p75NTR in the absence of ligand, by altering its dimeric conformation state was investigated along with the effects on the downstream signaling of the receptor26. Using cysteine-scanning mutagenesis, a collection of constitutively active p75NTR mutants were generated for which their ability of being further activated by proBDNF was tested. HEK293 cells transfected with p75NTR mutants were treated with Recombinant human proBDNF protein (#B-257) and the effects on activated JNK stress-activated MAPK and caspase 3 were monitored as they are cellular components activated in the apoptosis pathway. Both JNK and caspase 3 could not be further activated by the addition of proBDNF, strongly suggesting that the mutations inserted are truly activating and do so in a ligand independent fashion24.
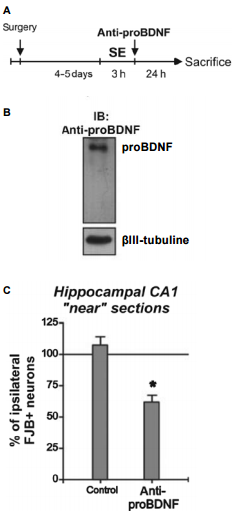
A) Animals received the administration of Anti-proBDNF Antibody (#ANT-006) after status epilepticus (SE) and neuronal damage was compared between hemispheres 24 hours later. B) Western blot analysis showing that the Anti-proBDNF administered detects a unique band corresponding to proBDNF from rat hippocampus. C) The administration of Anti-proBDNF after SE decreased the number of FJB+ neurons in the infused side to ~50%. Mean + SEM are indicated. Asterisk indicates significant differences compared with control animals. P < 0.05.Adapted from reference 25 with permission of Blackwell Publishing Ltd.
References
- Al-Shawi, R. et al. (2008) Eur. J. Neurosci. 27, 2103.
- Beattie, M.S. et al. (2002) Neuron 36, 375.
- Besser, M. and Wank, R. (1999) J. Immunol. 162, 6303.
- Bruno, M.A. et al. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 6735.
- Covolan, L. et al. (2000) Epilepsy Res. 39, 133.
- Fahnestock, M. et al. (2001) Mol. Cell. Neurosci. 18, 210.
- Fahnestock, M. et al. (2004) J. Neurochem. 89, 581.
- Fauchais, A.L. et al. (2008) J. Immunol. 181, 3027.
- Forge, A. and Schacht, J. (2000) Audiol. Neurootol. 5, 3.
- Fujikawa, D.G. et al. (1996) Brain Res. 725, 11.
- Jansen, P. et al. (2007) Nat. Neurosci. 10, 1449.
- Lee, R. et al. (2001) Science 294, 1945.
- Matsumoto, T. et al. (2008) Nat. Neurosci. 11, 131.
- Nagappan, G. et al. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 1267.
- Nykjaer, A. et al. (2004) Nature 427, 843.
- Pang, P.T. et al. (2004) Science 306, 487.
- Rattenholl, A. et al. (2001) J. Mol. Biol. 305, 523.
- Rudge, J.S. et al. (1998) Exp. Neurol. 149, 398.
- Schmidt-Kastner, R. et al. (1996) Exp. Brain Res. 107, 331.
- Schimmang, T. et al. (1995) Development 121, 3381.
- Suter, U. et al. (1991) EMBO J. 10, 2395.
- Tan, J. and Shepherd, R.K. (2006) Am. J. Pathol. 169, 528.
- Teng, H.K. et al. (2005) J. Neurosci. 25, 5455.
- Unsain, N. et al. (2008) Neuroscience 154, 978.
- Unsain, N. et al. (2009) J. Neurochem. 111, 428.
- Vilar, M. et al. (2009) J. Cell Sci. 122, 3351.
- Wang, Y.J. et al. (2009) Neurotox. Res. 15, 3.
- Woo, N.H. et al. (2005) Nat. Neurosci. 8, 1069.
- Yang, J. et al. (2009) Nat. Neurosci. 12, 113.
- Yune, T.Y. et al. (2007) J. Neurosci. 27, 7751.