Introduction
Voltage-gated calcium (CaV) channels play a major role in the normal functioning and pathophysiology of neurons and other excitable cells. Their role includes supply of Ca2+ for transmitter release, regulation of excitability by activation of Ca2+-dependent currents and activation of other Ca2+-dependent processes, including control of gene expression. Since Ca2+ entry regulates so many cellular processes, the correct trafficking and localization of CaV channels is of great importance for the normal functioning of cells.
Recently, mutations in a number of genes, causing disease in humans and mice, have been implicated in the context of voltage dependent ion channels and termed channelopathies1. Calcium channelopathies therefore refer to diseases caused by mutations in genes for the different proteins that together form a functional CaV channel. Here we review the known CaV channelopathies, which may appear due to mutations in any gene encoding α subunit that composes a functional CaV channel. In order to place these channelopathies in context, we shall also review the particular contribution of each subunit and their interactions within the channel complex.
Spontaneously arising channelopathies are also valuable as a research tool and together with the targeted gene-knockout studies2-10, this forms a basis for further understanding of the physiology involving these protein complexes, with their implications for neuronal and cardiovascular function and development. However, mutations in genes often lead to expression of a mutant or truncated protein. This in turn may either have an impact on the way the channel operates or influence the function of other proteins, either in a recessive or dominant manner.
1. Voltage-gated calcium channels: subunit structure and function
1.1 α1- The pore forming subunit of CaV channels
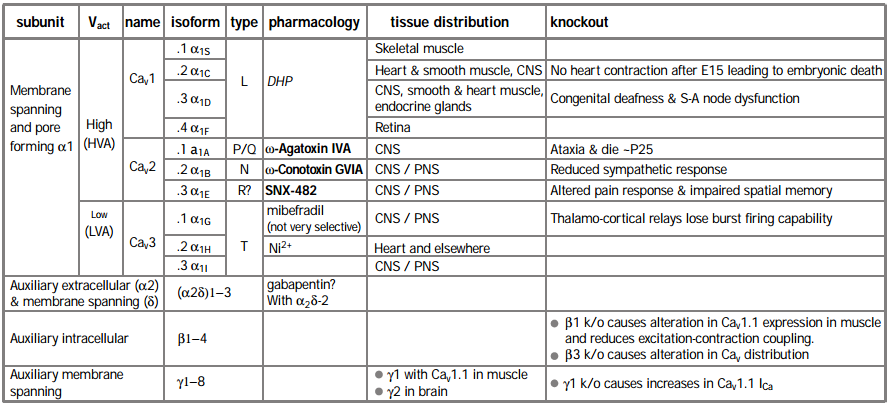
CaV channels consist primarily of a transmembrane α1 subunit, which has four domains of similar structure, each containing six putative transmembrane (TM) a helices and a re-entrant pore (P) loop between TM5 and 6, thought to form the lining of the ion-conducting pore (Figure 1). It appears that the α1 subunit folds up to form a donut-like structure that surrounds the central pore of the channel. This pore-forming α1 subunit contains also the cytoplasmic loops connecting the α1 domains, and intracellular N- and C-termini. Ten α1 subunits have now been cloned11 and six of them have been the subject of targeted gene knockout in mice (Table 1).
The L type CaV channels α1S (CaV1.1), α1C (CaV1.2) and α1D (CaV1.3) are all sensitive to 1,4-dihydropyridine (DHP) agonist and antagonist drugs. A fourth gene α1F (CaV1.4) was identified by virtue of the fact that patients with X-linked stationary night blindness carry a mutated form of the gene. CaV1.4 is expressed only in retina12,13. Its identification as an L type channel currently rests on sequence homology, as it has not yet been functionally expressed in a heterologous system. Mice lacking CaV1.2 die as embryos due to cardiac dysfunction beyond the age of E156. In contrast, knockout of another L-type channel, CaV1.3, resulted in viable mice with main deficiencies in sino-atrial node function in the heart and loss of hearing5.
The non-L type HVA CaV channels are encoded by α1A (CaV2.1) and α1B (CaV2.2) which respectively form ω-Agatoxin IVA-sensitive P/Q channels14-17 and ω-Conotoxin GVIA-sensitive N type channels18. Ablation of CaV2.1 results in mice with shorter life span and neurological abnormalities associated with ataxic phenotype, very similar to those seen in spontaneously arising CaV2.1 mutant mice4 (see below) stressing its role as a major neuronal CaV channel. Ablating the CaV2.2 gene in mice caused a less striking phenotype, with alterations in the sympathetic response, resulting in reduced blood pressure and enhanced heart rate9. The functional counterpart of the α1E (CaV2.3) channel remains unclear. It was initially suggested to form a subtype of mid – low voltage-activated “T” type channels19. The α1E channel has also been suggested to encode residual (R type) HVA channels that are insensitive to the blockers of L, N and P/Q channels20, although such a definition relies on complete block by these antagonists. However, a toxin (SNX-482 from Tarantula Hysterocrates gigas) that blocks α1E channels only blocks a small proportion of R type current in several systems21,22. Furthermore, α1E knockout mice retain a large proportion of R type current7. They also retain LTP and fear memory but show a deficiency in spatial memory23 and show altered pain responses24. Thus the physiological counterpart of α1E channels is still uncertain. Recently, three novel α1 subunit genes (α1G, H and I, or CaV3.1-3.3) have been cloned, encoding low voltage activated (LVA) T type channels25-27. CaV3.1 knockout mice are viable and fertile but lack burst firing in thalamocortical neurons10, confirming the role of this channel in the generation of action potential bursting28.
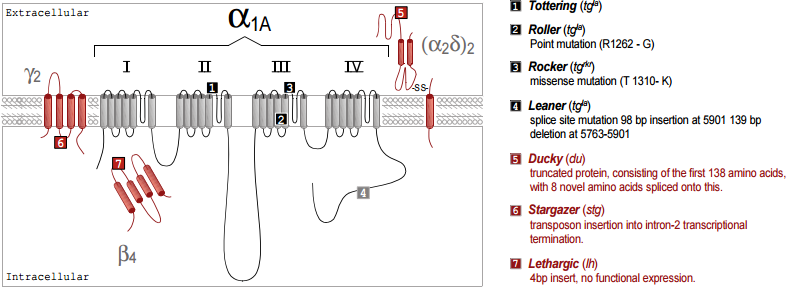
* In a functional HVA channel (and possibly LVA), the α1 subunit interacts with auxiliary subunits of the type: α1x/(α2δ)x /βx, γx may also be associated.
1.2 Auxiliary subunits in CaV channels
For those high-voltage activated (HVA) CaV channels where purification studies have been performed, the α1 subunit has been found to co-purify with an intracellular b subunit and an extracellular α2 subunit, which is attached by S-S bonds to a transmembrane subunit29-32 (Figure 1). The β subunit influences many of the channel properties including open time, activation, inactivation and modulation by other proteins (for example, see footnote 33. For review see footnote 34). However, one of the most apparent roles of all the β subunits is to increase membrane expression of the α1 subunit. This is postulated to occur by binding to an endoplasmic reticulum retention signal, allowing the channel to move out of this intracellular compartment35. Knockout of the skeletal muscle β1 isoform resulted in reduced expression of CaV1.1 in myotubes and impairment in excitation-contraction coupling2 and knockout of the β3 isoform resulted in an altered balance between the CaV channel types that were active in neurons and in altered kinetics of some CaV subtypes3.
Skeletal muscle CaV channels also co-purify with a γ subunit (γ1)36, and this knockout results in an increase in CaV channel currents in myotubes8. Whether the recently cloned novel γ-like subunits (γ2 – 8) are tightly associated with other types of CaV channels remains to be determined37-39. Certainly, in the original purification studies of N and P/Q CaV channels, no proteins of the molecular weights expected of γ subunits were identified32,40,41, although stargazin or γ2 (and a similar γ3) have recently been co-immunoprecipitated from brain tissue with CaV2.1 and CaV2.242. This study also showed that γ subunits reduced expression of CaV channels. Nevertheless, it is known that γ2 plays an important functional role in neurons, as its loss in the stargazer mutant mouse leads to a phenotype of spike and wave epilepsy37. However stargazer mice, and the allele waggler also lose cell surface expression of the AMPA glutamate receptors on cerebellar granule cells43,44. This observation has led to the discovery that γ2 is involved in trafficking AMPA receptors to postsynaptic membranes and that it also binds, via its four C terminal amino acids, to one of the PDZ domains in PSD -95 (a major Post Synaptic Density protein)45. Thus the neuronal γ subunits may have a more general role in regulating cell surface expression of a number of receptors and ion channels.
2. Calcium channel trafficking and disease
A number of human genetic diseases are associated with mutations in L- and P/Q-type CaV channels (Tables 1 and 2). Below, we review these diseases and also examine mutations in similar genes in mice that serve as models for epilepsy and ataxia.
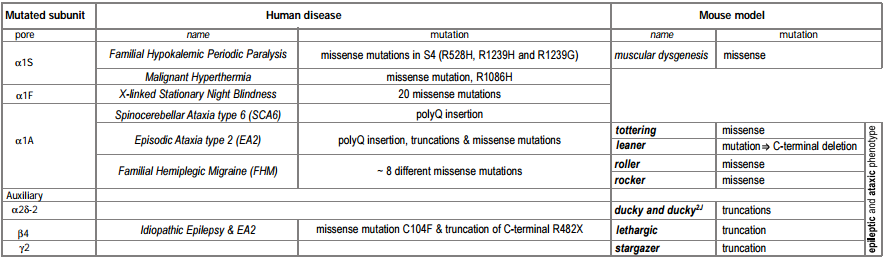
The genetic basis of a number of spontaneously occurring mouse mutants with absence epilepsy and cerebellar ataxia has been identified to involve mutations in CaV channel subunits. These include tottering, leaner, roller and rocker, which have mutations in the pore forming CaV 2.1 (α1A)46-49. Mutated auxiliary subunits include the following: stargazer, which has a truncation mutation in γ237, lethargic which has a truncation mutation in β450 and ducky, which has a truncation mutation in α2δ-251. It is interesting that four out of five mouse mutations established as models for absence epilepsy occur in genes encoding all the known components of a CaV channel heteromultimer (Figure 1). Some of these mutations also affect synaptic transmission52, an effect which may be very selective, influencing excitatory but not inhibitory transmission in the thalamus53.
Our results concerning expression and targeting in Madin Darby Canine Kidney (MDCK) cells54 suggest that auxiliary β subunits can exert an effect on the targeting of the CaV channel complex, particularly for the CaV2.1 subunit. This provides a possible mechanism for the major cerebellar deficit found in the lethargic mutant mouse, which has a mutation that results in a truncated β4 subunit message and no β4 full length protein50. Since, in expression studies, all β subunits are able to interact with the CaV2.1 subunit55,56, until now it has been unclear how the absence only of β4 could produce the symptoms experienced by this mouse mutant. However, extrapolating from our results in MDCK cells, β4 may be one of only two β subunits able to target CaV2.1 to presynaptic sites, and the other, β1b, has low expression in the adult brain57,58. It is therefore quite conceivable that the loss of β4 could produce aberrant targeting of CaV2.1, and result in major defects in cerebellar development and function. It has also been shown that β1b levels are increased in lethargic mice, which may be an adaptive developmental response to the lack of β458.
It was shown recently that the spontaneously arising mouse mutant, ducky, which has absence epilepsy and ataxia, due to a mutation in α2δ-2, does produce a truncated protein, consisting of the first 138 amino acids of the protein, with 8 novel amino acids spliced onto this. The α2γ-2 protein is normally expressed strongly and selectively in Purkinje cells in the cerebellum, both in the dendrites and the cell bodies. The CaV channel currents are markedly reduced in the homozygous ducky Purkinje cells51 and expression of the mutant α2δ-2 protein may contribute to this phenotype59.
It is notable that the most prevalent CaV channel subunit associated with neurological disorders is CaV2.1 (for review see footnote 60). This is likely to be a result of the important role played by CaV2.1 in synaptic transmission (for review see footnote 61), but also reflects the fact that it is highly prevalent in the cerebellum. An ataxic phenotype is often correlated with abnormal development or degeneration of the cerebellum (for example see footnote 62). Therefore the mutated CaV component may lead to an imbalance of calcium homeostasis that causes abnormal neuronal functioning and may eventually result in apoptosis63. When such a mutation is in a gene that is strongly expressed in the cerebellum, like CaV2.1, β4 or α2δ-2, it is likely to lead to cerebellar degeneration and movement disorders64.
2.2. Human diseases resulting from mutations in CaV components
2.2.1. Human L-type channelopathies
Several muscular and sight-related diseases in humans have been found to involve mutations in L-type CaV channels. These include hypokalemic periodic paralysis and malignant hyperthermia, which involve, respectively, several or single mutations in CaV1.166,67 and congenital stationary night blindness, resulting from ~20 different point mutations in the retina-specific CaV channel CaV1.4 (α1F)12, 68.
2.2.2. Human P/Q-type channelopathies
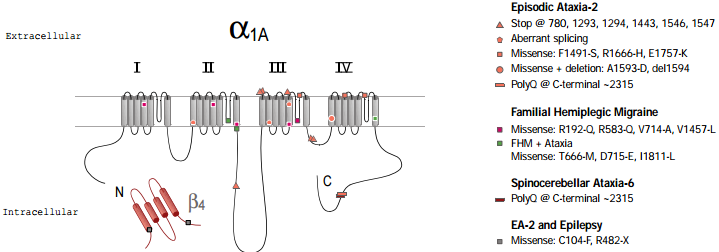
Three different, but related, human diseases arise from mutations in the CaV2.1 (α1A) subunit gene (figure 2). These include familial hemiplegic migraine (FHM) which results from point mutations69, episodic ataxia type 2 (EA2), which is associated mainly with a variety of truncation mutations69,70 and spinocerebellar ataxia (SCA6), which involves the expansion of a CAG repeat in one particular splice variant of α1A71,72. A number of the disease phenotypes may result in part from defects in trafficking. For example in SCA6, a dominant disease, the CAG expansion is translated as a polyglutamine repeat in the C terminus of a particular splice variant of α1A. Although the expansion is smaller than in other polyglutamine diseases, it has been suggested to result in cytoplasmic aggregations of the protein in Purkinje cells of SCA6 patients71. Furthermore, SCA6 mutant CaV2.1 constructs, with CAG repeats of sufficient length to cause disease, were found as perinuclear aggregations when the mutant channels were expressed in HEK293 cells71, although in this study no accessory β subunits were co-expressed with the channels. In this view abnormal trafficking and therefore aggregation of CaV2.1 caused the disease. An alternative view is that the polyglutamine repeat affects CaV channel properties, particularly when present in the P type splice variant of CaV2.1 (which is missing two amino acids, NP, in an extracellular loop of domain IV) that is highly prevalent in Purkinje cells72. In EA2, which is also a dominant disease, most of the truncation mutations occur after the end of domain II70, so that substantial portions of the CaV2.1 channel may be synthesized and inserted into the plasma membrane.
However, missense73,74 mutations and a poly-Q insertion similar to the one found in SCA675, in EA2 patients have also been described. All the mutations related to EA2 that were examined showed complete or partial loss of function74. The truncated proteins may also have a dominant negative effect, so that the disease phenotype would be caused by a gain of function, rather than being solely due to a reduction in normal CaV2.1 being produced from the single wildtype copy of the gene.
It is possible that such a dominant negative effect may be due to interference with the correct folding, trafficking or functioning of the wildtype CaV2.1 channels. Indeed, we have recently shown that truncated constructs of CaV2.2 have a dominant negative effect on the expression of CaV2.276. It is of interest that in the purification studies of CaV2.1 and CaV2.2, a substantial protein band of about 95 kD was always observed32,41, which was identified by microsequencing as the first two domains of CaV2.177. It is unknown whether this protein is a breakdown product of full length CaV2.1, in which case it must be very stable, or whether its synthesis involves premature truncation of the nascent protein. One might expect that if the EA2 mutant proteins have a dominant negative effect, that this might also be the case for this 95 kD protein.
Familial hemiplegic migraine also involves mutations in the CaV2.1 gene, but in contrast to the EA2 mutations, these are all point missense mutations leading to either partial loss of function or even to increased activity of the channels in some mutations78,79. All three diseases (SCA6, EA2 and FHM) arise from mutations in the same gene and show an overlapping spectrum of symptoms, with phenotypes often involving ataxia. The severity of the phenotype appears to correlate with those mutations that cause either the loss of function of the channel or toxicity, which is presumed to occur from aggregation of a mutated or truncated product. Each of these mutations will affect calcium homeostasis and cell toxicity to a different degree. Therefore, the cumulative effect, resulting in cell death and clinical symptoms, will manifest itself at different times and with different impact.
In addition to mutations in the CaV2.1, gene, mutations in the β4 subunit were found in patients with idiopathic generalized epilepsy and episodic ataxia80. The similarity of symptoms reinforces the view that these two subunits interact physiologically.
One must bear in mind that many mutations in other voltage- and ligand- gated channels result in similar phenotypes and disease1. This is the case in malignant hyperthermia in which in addition to CaV1.1, mutations in the ryanodine receptor (RyR1), which links functionally with CaV1.1 in excitation-contraction coupling can also cause the same disease. The same applies to mutations in sodium and potassium channels, which cause diseases with ataxic phenotypes similar to mutations in neuronal CaV components81,82. These examples reinforce the view that a specific and accurate electrochemical balance is required for neuronal functioning, to both acquire its architecture during development and to function normally. Therefore, in a given cell, many channelopathies may result in degeneration, and the symptoms will depend on the localization and severity of the electrochemical imbalance.
* See1, 65 for reviews. For specific references see text.
Acknowledgments to Annette C. Dolphin, Professor of Pharmacology, Department of Pharmacology, University College London (UCL), London UK. This work was supported by the Wellcome Trust.
References
- Lehmann-Horn, F. & Jurkat-Rott K. (1999) Physiological Reviews 79, 1317-72.
- Gregg, R. G. et al. (1996) P.N.A.S. USA 93 (24), 13961-6.
- Namkung, Y. et al. (1998) P.N.A.S. USA 95 (20), 12010-15.
- Jun, K. et al. (1999) P.N.A.S. USA 96 (26), 15245-50.
- Platzer, J. et al. (2000) Cell 102 (1), 89-97.
- Seisenberger, C. et al. (2000) J. Biol. Chem. 275 (50), 39193-9.
- Wilson, S.M. et al. (2000) J. Neurosci. 20, 8566-8571.
- Freise, D. et al. (2000) J. Biol. Chem. 275 (19), 14476-81.
- Ino, M. et al. (2001) P.N.A.S. USA 98 (9), 5323-8.
- Kim, D. et al. (2001) Neuron 31, 35-45.
- Ertel, E.A. et al. (2000) Neuron 25, 533-535.
- Bech-Hansen, N. T. et al. (1998) Nature Genetics 19, 264-267.
- Strom, T. M. et al. (1998) Nature Genetics 19, 260-263.
- Sather, W. A. et al. (1993) Neuron 11, 291-303.
- Gillard, S. E. et al. (1997) Neuropharmacology 36, 405-409.
- Berrow, N.S. et al. (1997) European Journal of Neuroscience 9, 739-748.
- Bourinet, E. et al. (1999) Nature Neuroscience 2, 407-415.
- Fujita, Y. et al. (1993) Neuron 10, 585-598.
- Soong, T.W. et al. (1993) Science 260, 1133-1136.
- Randall, A. & Tsien R.W. (1995) J. Neurosci. 15, 2995-3012.
- Newcomb, R. et al. (1998) Biochemistry 37, 15353-15362.
- Tottene, A. et al. (2000) J. Neurosci. 20, 171-178.
- Kubota, M. et al. (2001) Biochem. Biophys. Res. Comm. 282, 242-8.
- Saegusa, H. et al. (2001) P.N.A.S. USA 97 (11), 6132-7.
- Perez-Reyes, E. et al. (1998) Nature 391, 896-900.
- Cribbs, L.L. et al. (1998) Circulation Research 83, 103-109.
- Lee, J. H. et al. (1999) J. Neurosci. 19, 1912-1921.
- Huguenard, J. R. & Prince D. A. (1994) J. Neurosci 14, 5485-5502.
- Tanabe, T. et al. (1987) Nature 328, 313-318.
- Chang, F.C. & Hosey M.M. (1988) J. Biol. Chem. 263, 18929-18937.
- Witcher, D.R. et al. (1993) Science 261, 486-489.
- Liu, H. et al. (1996) J. Biol. Chem. 271, 13804-13810.
- Meir, A. et al. (2000) Biophys. J. 79 (2), 731-746.
- Walker, D. & De Waard M. (1998) Tins 21, 148-154.
- Bichet, D. et al. (2000) Neuron 25, 177-190.
- Takahashi, M. et al. (1987) P.N.A.S. USA 84, 5478-5482.
- Letts, V.A. et al. (1998) Nature Genetics 19, 340-347.
- Black, J.L. & Lennon V.A. (1999) Mayo Clinic Proceedings 74, 357-361.
- Klugbauer, N. et al. (2000) FEBS Letters 470, 189-197.
- McEnery, M.W. et al. (1991) P.N.A.S. USA 88, 11095-11099.
- Witcher, D.R. et al. (1994) Methods in Enzymology 238, 335-348.
- Kang, M. G. et al. (2001) J. Biol. Chem. (July 5th) in press.
- Hashimoto, K. et al. (1999) J. Neurosci. 19, 6027-6036.
- Chen, L. et al. (1999) P.N.A.S. USA 96, 12132-12137.
- Chetkovich, D.M. et al. (2000) Soc. Neurosci. Abstracts 26, 717.5
- Fletcher, C.F. et al. (1996) Cell 87, 607-617.
- Doyle, J. et al. (1997) Mamm. Genome 8, 113-120.
- Mori, Y. et al. (2000) J. Neurosci. 20 (15), 5654-5662.
- Zwingman, T. A. et al. (2001) J Neurosci. 21 (4), 1169-78.
- Burgess, D.L. et al. (1997) Cell 88, 385-392.
- Barclay, J. et al. (2001) J. Neurosci. 21 (16), 6095-6104.
- Qian, J. & Noebels J. L. (2000) J. Neurosci. 20 (1), 163-170.
- Caddick, S. J. et al. (1999) J. Neurophysiol. 81, 2066-2074.
- Brice, N.L. & Dolphin A.C. (1999) J. Physiol. 515, 685-694.
- De Waard, M. & Campbell, K.P. (1995) J. Physiol. 485, 619-634.
- Brice, N.L. et al. (1997) European J. Neurosci. 9, 749-759.
- Ludwig, A. et al. (1997) J. Neurosci. 17, 1339-1349.
- McEnery, M.W. V. (1998) J. Biol. Chem. 273, 21435-21438.
- Brodbeck, J. et al. (2002) J. Biol. Chem. 277, 7684.
- Jen, J. (1999) Current Opinion in Neurobiology 9, 274-280.
- Meir, A. et al. (1999) Physiological Reviews 79 (3), 1019-88.
- Fletcher, C. F. et al. (2001) FASEB J 15 (7), 1288-1290.
- Herrup, K. & Wilczynski S. L. (1982) Neuroscience 7, 2185-2196.
- Tanaka, O. et al. (1995) Brain Res. Mol. Brain Res. 30, 1-16.
- Burgess, D. L & Noebels, J. L. (1999) Ann. NY Acad. Sci. 868, 19-212.
- Jurkat-Rott, K. et al. (1994) Hum. Mol. Genet. 3 (8), 1415-1419.
- Monier, N. et al. (1997) Am. J. Hum. Genet. 60 (6), 1316-1325.
- Boycott, K. M. et al. (2001) Hum. Genet. 108, 91-97.
- Ophoff, R.A. et al. (1996) Cell 87, 543-552.
- Denier, C. et al. (1999) Neurology 52, 1816-1821.
- Ishikawa, K. et al. (1999) Hum. Mol. Genet. 8, 1185-1193.
- Toru, S. et al. (2000) J. Biol. Chem. 275, 10893-10898.
- Friend, K. L. et al. (1999) Hum. Genet. 105, 261-265.
- Guida, S. et al. (2001) Am. J. Hum. Genet. 68, 759-64.
- Jodice, C. et al. (1997) Hum. Mol. Genet. 6 (11), 1973-1978.
- Raghib, A., et al. (2001) J. Neurosci. 21, 8495-8504.
- Scott, V.E.S. et al. (1998) J. Neurosci. 18, 641-647.
- Kraus, R. L. et al. (2000) J. Biol. Chem. 275 (13), 9239-43.
- Hans, M. et al. (1999) J. Neurosci. 19 (5), 1610-19.
- Escayg, A. et al. (2000) Am. J. Hum. Genet. 66, 1531-39.
- Kohrman, D. C. et al. (1996) J. Neurosci. 16, 5993-5999.
- Lit M. et al. (1994) Am. J. Human. Genet. 55, 702-709.