Tetrodotoxin (TTX) is an organic molecule that is very toxic to mammals and many other organisms. It is produced by certain species of bacteria and is accumulated in organs of organisms which consume it via the food chain. Tetrodotoxin is most commonly found in the puffer fish (tetradontidae) but can also be isolated from different species of frog, salamandra, octopus and star fish. The toxicity of Tetrodotoxin is derived from its high affinity, and interaction with some of the most abundant voltage dependent Na+ (NaV) channels, leading to its ability to inhibit electrical activity in nerve and muscle. Below, we describe recent use of Alomone Labs Tetrodotoxin (#T-500) and Tetrodotoxin citrate (#T-550) in several types of experiments ranging from brain slice recordings to in vivo secretion measurement.
Introduction
NaV channels are plasma membrane proteins which upon their activation by voltage drops in the plasma membrane potential, generate a Na+ selective pore and allow for a few milliseconds the passage of Na+ ions into the cell. This inflow of positively charged ions further decreases the voltage across the membrane causing activation of more NaV channels as well as KV channels (allowing K+ ions outflow and restorating the resting membrane potential). This mechanism is at the basis of action potential (AP) generation and propagation in neurons and some muscle cells. All neuronal activity and communication is encoded or derived from APs traveling along the membrane of neurons which also affect synaptic transmission that pass such encoded information to a neighboring cell. Inhibition of AP completely halts electrical activity and dependent mechanisms in excitable cells.
Tetrodotoxin was first discovered as a specific NaV channel blocker by Narahashi in the 1960s where he showed that it blocks the frog muscle AP. Later, using voltage clamp it was shown that TTX blocks the Na+ conductance without affecting K+ conductance in a giant lobster nerve axon. Since, TTX is considered to be a very specific and potent pharmacological tool and is one of the first defined ion channel blockers. The use of TTX has also led to the notion that some APs are resistant to TTX block. These are generated by activation of either NaV TTX-resistant channels or by low voltage activated CaV channels. The best known examples of TTX resistant APs are in the heart (where the TTX-resistant NaV1.5 is the main isoform) and in some sensory DRG neurons (where NaV1.8 and NaV1.9 along with TTX-sensitive channels are expressed)63.
In mammals, TTX potently blocks (IC50 in the tens of nanomolar range) a subfamily of NaV channels, including NaV1.1, 1.2, 1.3, 1.6 and 1.7 in the central nervous system and NaV1.4 in skeletal muscle. Figure 1 shows a complete block of NaV currents in cultured rat neocortical neurons118. The potent effect of TTX on AP propagation (which depends on NaV channel activity in axons) was also demonstrated in different types of nerve fibers from dorsal root ganglion (DRG) neurons. An example of the effect of low dosage of TTX on C-fiber conduction74 is shown in Figure 2.
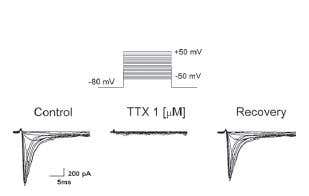
Figure 1. Voltage-Dependent Inward Currents Are Blocked by Tetrodotoxin in Cultured Rat Neocortical Cells. Depolarizing steps from −50 mV to −5 mV, step 5 mV, from a holding potential of −80 mV, induced in low concentration Na+ solution (10 mM) inward currents that were blocked by the perfusion of 1μM Tetrodotoxin citrate (#T-550). The neuron was 8 days old.
Adapted from reference 118 with permission of Elsevier.
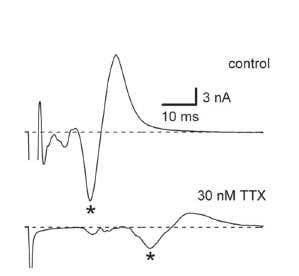
Figure 2. Slowing of C-Fiber Conduction by Tetrodotoxin. A) C-fiber compound action potential currents recorded in a caudal dorsal root in the presence and absence of 30 nM Tetrodotoxin or Tetrodotoxin citrate (#T-500 or #T-550, respectively). B) Latency of the negative wave peak as a function of Tetrodotoxin concentration. Latencies of C-waves were normalized to those in control (IC50 = 26 ±2 nM, n = 6).
Adapted from reference 74 with permission of The American Physiological Society.
Tetrodotoxin inhibits NaV channels by binding to the narrowest part of the Na+ channel pore (i.e. the selectivity filter). By doing so, it prevents Na+ ions from binding and permeating the cell via the open pore. The TTX binding site in the selectivity filter is complex and involves a few amino acids from each NaV channel domain. TTX-resistant NaV channels have a one amino acid difference in this region, which reduces the affinity to TTX by over 1000 (for review see reference 26). In a similar manner it was recently shown that animals, like the puffer fish, which accumulate TTX have a similar point mutation in their NaV channels making them resistant to TTX intoxication (for review see reference 95). The selectivity of TTX to NaV versus other voltage dependent channels makes it an ideal pharmacological tool in assessing the roles of NaV channels, APs and neuronal activity in different experimental settings.
The Use of Tetrodotoxin in the Laboratory
Whenever one wants to shut down neuronal or muscular electrical activity, to create a less noisy electrical environment or to isolate the effect of such activity, TTX is usually the first choice.
In mammalian brain slice elctrophysiological recordings, especially when synaptic mechanisms are studied, TTX is often used (usually with some transmitter gated channel blocker) to eliminate any electrical input and facilitate the monitoring of the neuron’s spontaneous activity. Miniature excitatory or inhibitory synaptic currents or potentials (mEPSC and mIPSC respectively) are used to discriminate between presynaptic and postsynaptic mechanisms of action. In order to record these tiny currents, many investigators use TTX and examine the effects of compounds or treatments on the synapse activity6‑8,11,14,15,19‑21,29,32,36‑39,41,43‑45,48,49,51,52,54‑56,60,68,71,78,80,81,83‑85,89,91,96,98‑102,104,106,108,109,113‑116.
Tetrodotoxin is also used in synaptic recordings to assess the role of NaV channel activity in complex synaptic feedback mechanisms. For example, in the retina, application of TTX does not directly affect transmitter release from most retinal bipolar cells (RBC) but abolishes lateral feedback transmission onto RBCs. TTX also eliminates both responses to CPPG and kainate, confirming that both ON and/or OFF-responding GABAergic amacrine cells providing lateral feedback rely heavily on NaV-dependent signaling12,13. In some cases, the shutdown of electrical activity is not only a means, but a treatment to be studied. This is nicely demonstrated in a study that examined the influence of neuronal network activity on synapse dynamics by using long term culture and monitoring a fluorescent synaptic marker. TTX was used to diminish network activity and was found to influence synapse remodeling59. TTX-sensitive NaV channel-dependent electrical activity was also shown to partially affect the location of the axon initial segment of neurons and such a form of activity-dependent plasticity may fine tune neuronal excitability during development30.
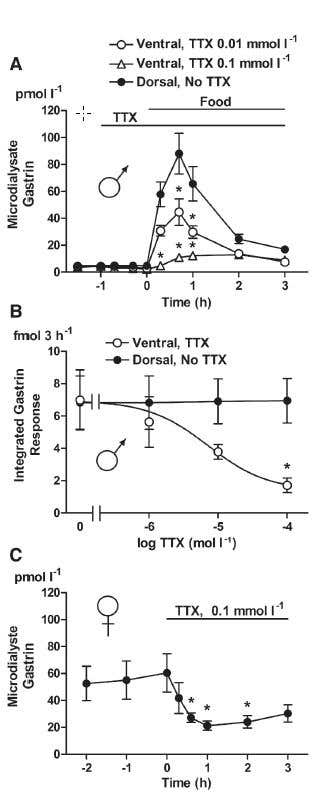
Figure 3. Gastrin Mobilization in Response to Food Intake during Local Infusion of Tetrodotoxin. A) Gastrin in microdialysate was monitored in response to food intake in intact, fasting male rats equipped with microdialysis probes on both the ventral (○, Δ) and the dorsal (●) sides of the antrum. Access to food is indicated. Local infusion of 10 μM Tetrodotoxin citrate (#T-550), (○, n = 7) or 100 μM Tetrodotoxin (with citrate), (Δ, n = 6) via the ventral microdialysis probe started 1 h before the start of food intake and continued throughout the experiment (as indicated). B) Integrated microdialysate gastrin response on the TTX-treated, ventral side and on the non-treated, dorsal side during 3 h of food intake (n = 4-8). C) Gastrin in microdialysate was monitored in response to local infusion of Tetrodotoxin (with citrate) in omeprazole treated.
Adapted from reference 22 with permission of Elsevier.
Synaptic transmission and secretion are often studied by directly measuring transmitter or hormone quantities in the extracellular solution or in vivo. Using TTX, it was demonstrated that dopamine release from nucleus accumulus neurons is dependent on electrical activity24. GABA and glutamate release in response to pyrethroid insecticides in vivo was totally abolished by TTX administration33. Measuring gastrin in the antrum in vivo demonstrated that food intake initiates release which is dependent on neuronal activity and is inhibited by TTX22 (Figure 3). The dependence of secretion on NaV channels is nicely demonstrated in the pancreas, where TTX potently blocks insulin release from beta cells. In a NaV channel β1 subunit mutant, insulin secretion from beta cells is altered, but still TTX prevents secretion23.
Tetrodotoxin is often used in nerve muscle and gland preparations to discriminate between processes originating directly from the muscle and those originating from a nerve innervating the muscle34,35,47,92,103,105,107. In the mouse ileal longitudinal muscle, a selective CB1 receptor agonist (ACEA) reduced spontaneous contractions. This effect was almost abolished by TTX. In addition, contractile responses to α, β-MeATP (P2X receptor agonist) were virtually abolished by TTX suggesting a neuronal control of these modulatory actions4. In skeletal muscle, the TTX-sensitive channel activity was shown to be important for normal development via Syndecan-4 and β1 integrin97 (Figure 4).
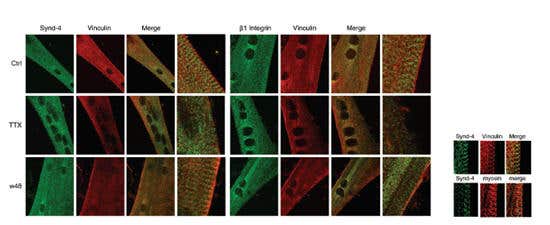
Figure 4. Co-Localization of Syndecan-4 and β1 Integrin with Vinculin Is Disrupted in Tetrodotoxin-Treated Myotubes. Left and right panels, indirect immunofluorescence images showing the sub-cellular localization of both syndecan-4 and β1 integrin with vinculin in control cultures at day 5 of differentiation. The left and right columns in each panel correspond to merged images matching to high magnification selected areas of corresponding images. Scale bar, 2.5 μm). Middle rows in each panel, indirect immunofluorescence images showing the disrupted sub-cellular localization of both syndecan-4 and β1 integrin with vinculin in 1 μM Tetrodotoxin citrate (#T-550) treated cultures at day 5 of differentiation. Lower rows in each panel, indirect immunofluorescence images of Tetrodotoxin (with citrate)-treated cultures for 2 days, washed out and fixed 48h afterwards (w48), showing the recovery of the co-localization of both syndecan-4 and β1 integrin with vinculin. Scale bar, 10 μm. The inset shows indirect immunofluorescence images at high magnification: the sub-cellular co-localization of syndecan-4 and vinculin between the myosin heavy chain in a control myotube at day 4 of differentiation. Scale bar, 1.5 μm.
Adapted from reference 97 with permission of Elsevier.
Tetrodotoxin was used to demonstrate the contribution of NaV channels in dendrites to electrical activity and neuronal AP. In medial nucleus trapezoid body (MNTB) APs were recorded in a neuron before and during application of TTX to either the dendrite or the axon. Blocking local NaV channels in dendrites attenuated AP in the soma suggesting a regulatory role for these channels in neuronal repetitive AP firing50. In dendrites of neonatal lateral superior olive neurons, TTX application attenuated a dendritic Ca2+ signal, indicating the involvement of backpropagating AP46 (Figure 5). In another work it was demonstrated that nicotinic α7-derived Ca2+ transient in dendrites in hippocampal interneurons was mildly affected by TTX86. TTX was also shown to influence “noise” in dendrites implying that NaV channels contribute to electrical stability112.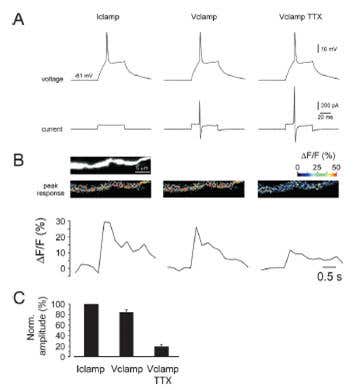
Figure 5. Global Dendritic Ca2+ Responses Depend on Back-Propagating Na+ Action Potentials in Neonatal Lateral Superior Olive Neurons. A) Stimulation protocol: Left: AP was first elicited in current clamp by a short depolarizing current injection. Middle: The voltage trace from left was used as command potential during voltage clamp. Right: The voltage clamp command is repeated after blocking Na+ channels with 1 μM Tetrodotoxin citrate (#T-550). B) Dendritic Ca2+ responses in the three conditions shown in A. C) Summary data from six neurons.
Adapted from reference 46 with permission of Elsevier.
In a series of experiments, the group of Djamgoz demonstrated the expression of NaV channels in malignant cell lines. In order to examine the contribution of these channels to transformation related cellular behaviors, TTX was used and its effect on processes ranging from cellular adhesion to migration capabilities was examined18,27,65,67. For example, the role of NaV channels in adhesion properties of MDA-MB-231 human breast cancer cells or the increased motility of Mat-LyLu rat prostate cancer cells following stimulation with epidermal growth factor (EGF) was demonstrated using TTX.
Influences of NaV channel activity on the expression of proteins and cellular processes are also studied in neurons. In DRG neurons it was demonstrated in vivo that inflammatory injury caused by carrageenan rapidly induced the expression of TNF-R1. This expression was relatively transient and TTX was able to counteract this expression leading to hyperexcitability and pain17.
Electrical activity in the form of AP also affects neuronal survival. In dissociated cultures of cortical neurons exposed to TTX for long periods of time, it was demonstrated that such activity block leads to the death of neurons but leaves glia intact. Culturing these cells with TTX eventually leads to increased apoptosis and death88.
Tetrodotoxin has been used to demonstrate neuronal differentiation of stem cells as AP firing (arising from NaV channel expression) is a phenotypic marker of neurons3,42,79. In one example, mouse inner ear stem cells were differentiated into sensory neurons and exhibited TTX sensitive inward currents as well as AP firing58 (Figure 6).
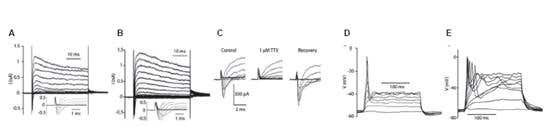
Figure 6. Differentiated Inner Ear Stem Cells Develop Electrical Features of Neurons. IV relations of an isolated cultured spiral ganglion neuron (A) and a cultured differentiated stem cell (B). Voltage clamp recordings from a holding potential of -84 mV, voltage steps to -104 to +36 mV in 10 mV increments. A slowly inactivating outward current and a rapidly activating and inactivating inward current were observed at about -34 mV or more positive (A and B, insets; traces for voltage steps from -84 to +16 mV are shown). C. The rapid inward current in differentiated stem cells was completely and reversibly inhibited by 1 μM Tetrodotoxin citrate (#T-550) indicating that it is a voltage gated Na+ current (INa). In current clamp, depolarizing current injections (10-120 pA in 10 pA increments) evoked action potentials in a cultured spiral ganglion neuron (D) and in a differentiated neuron cell (E).
Adapted from reference 58 with permission of John Wiley & Sons Ltd.
Changes in the expression of NaV channels play a role in many types of neuronal diseases such as neuropathic pain and epilepsy. Increased persistent NaV current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis (ALS). This was demonstrated by using TTX to identify differential expression between wild type and mutated neurons73.
While recording cellular ionic currents, it is sometimes necessary to eliminate pharmacologically “unwanted” compounds. For example, while recording Ca2+ channel activity in neurons, the activity of endogenous NaV channels might disrupt the interpretation of results, as these are also monitored as inward current. For such reasons TTX is often included in the extracellular solution in voltage clamp experiments (see examples in references 1, 9, 10, 31, 40, 53, 62, 64, 69, 70, 72, 75-77, 82, 93, 94, 110, 111,117). TTX is also often used to isolate the currents and effects of TTX-sensitive versus TTX-resistant channels in a variety of tissues such as DRG, heart and skeletal muscle (see for example references 5, 16, 57, 61, 74, 90), or simply to confirm the existence or contribution of NaV channels to a biological process2,25,28,66,87.