Roughly 60% of people with irritable bowel syndrome (IBS) also live with an anxiety disorder (1). Most clinicians have known this for decades, and most patients have lived with it for longer. And for almost as long, the working mental model has been that pain and anxiety in IBS sort of slosh around together – each feeding the other, indistinguishable in the brain because they feel indistinguishable in the body. Hence, the standard “vicious cycle” framing of pain, anxiety, more pain, more anxiety, and so on.
However, a paper in Neuron from Li et al., suggested that the brain has been keeping much more careful records of pain and anxiety (2).
A Model That Doesn't Cheat
Most studies of pain-anxiety comorbidity build the comorbidity by hand, where you induce visceral pain and then measure anxiety. Or you stress an adult animal, then ask whether it hurts. It’s useful, but you've inserted the answer into the design. What the field has been missing is a model where both phenotypes, pain and anxiety, arise naturally from the same upstream cause, the way they do in patients.
Li et al. used prenatal maternal stress (PMS) to study this. Pregnant mice were stressed, their offspring were raised normally, and the adult offspring spontaneously developed both visceral hypersensitivity (emerging at 6 weeks, persisting through 12) and anxiety-like behavior (still present at 16 weeks). The model had successfully reproduced the specific clinical phenotype, in both sexes, in animals that were never directly stressed.
That alone is a big step forward in studying these disorders, but the model also let them ask something the field has struggled to answer: in a brain handling both at once, which neurons are doing which job?
The Sorting Center
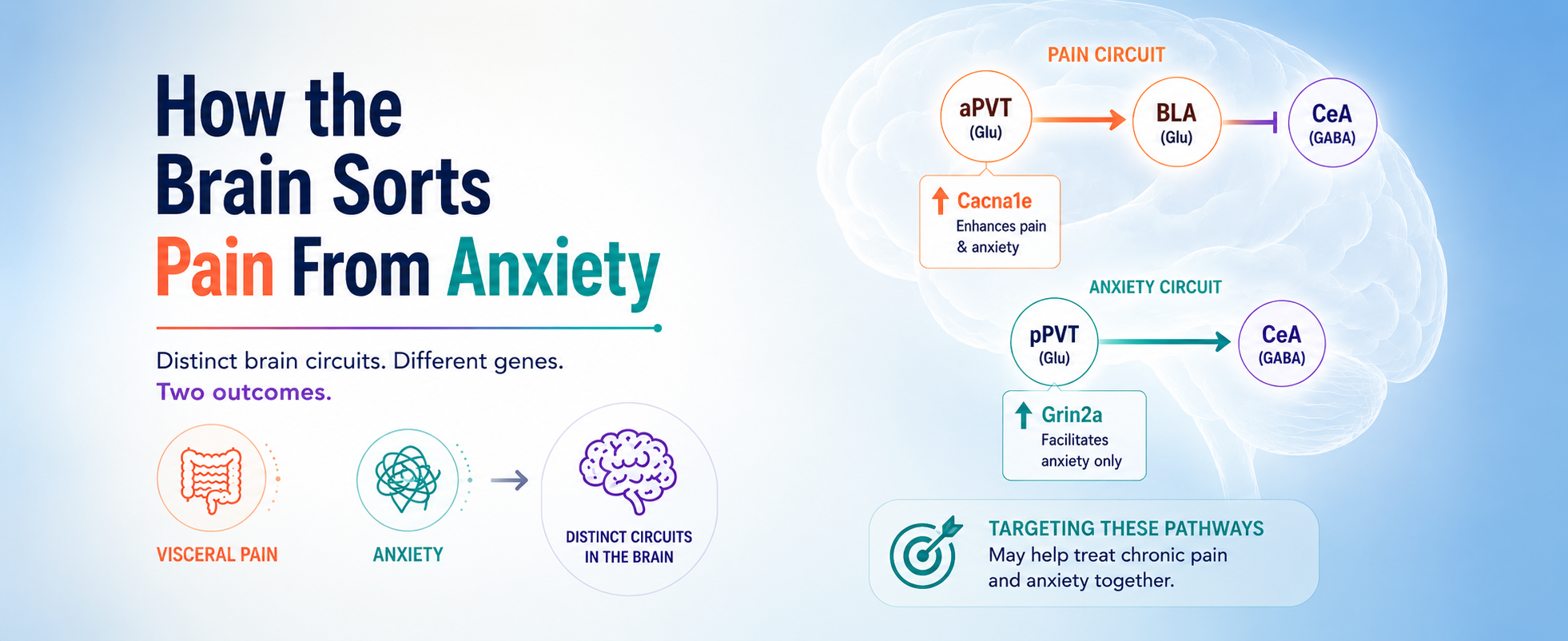
Searching for brain regions activated by colorectal distension (the pain stimulus) and elevated platform stress (the anxiety stimulus), the authors flagged the paraventricular thalamus (PVT) as showing the largest c-Fos (an immediate early gene that encodes a transcription factor, frequently used as a marker for neuronal activity) increase under both conditions. The results were not surprising since the PVT shows up in essentially every pain and emotion paper from the last decade.
Using a Tet-off labeling strategy that tags pain-activated neurons in one color and anxiety-activated neurons in another within the same mouse, they found that 82% of pain-responsive PVT neurons sit in the anterior PVT (aPVT), while 84% of anxiety-responsive neurons sit in the posterior PVT (pPVT) – with no overlap. Calcium imaging confirmed this result: aPVT glutamatergic neurons fire during colorectal distension and stay quiet during elevated platform stress; pPVT glutamatergic neurons do the opposite.
The PVT, traditionally treated as one nucleus, and described by the authors as “a sorting center that distinctly processes visceral pain and anxiety”, is running two operations on different shifts.
Two Genes, Two Subregions
RNA-seq analysis of the two subregions cleanly show the molecular split. The CaV2.3 R-type calcium channel gene (Cacna1e) is selectively upregulated in the aPVT of PMS mice. In contrast, the NMDA receptor GluN2A subunit gene (Grin2a), is selectively upregulated in the pPVT of PMS mice.
Immunofluorescence confirmed the protein expression pattern along the entire anterior-posterior axis. Using Alomone’s Anti-CaV2.3 (CACNA1E) Antibody (#ACC-006), the authors showed that the CACNA1E protein was specifically elevated in the aPVT, unchanged in the pPVT, and co-localized tightly with VGluT2 (Figure 1). Thus, the channel is upregulated exactly where the pain-responsive glutamatergic neurons reside. The pattern remains the same in both male and female PMS mice.
Knockdown studies revealed that short hairpin (sh)RNA directed against Cacna1e in aPVT glutamatergic neurons showed reduced calcium activity during colorectal distension and the visceral pain threshold was raised. Moreover, shRNA directed against Grin2a in pPVT glutamatergic neurons reduced their activity during stress and diminished anxiety-like behavior. Each gene does its own job, in its own subregion, full stop.
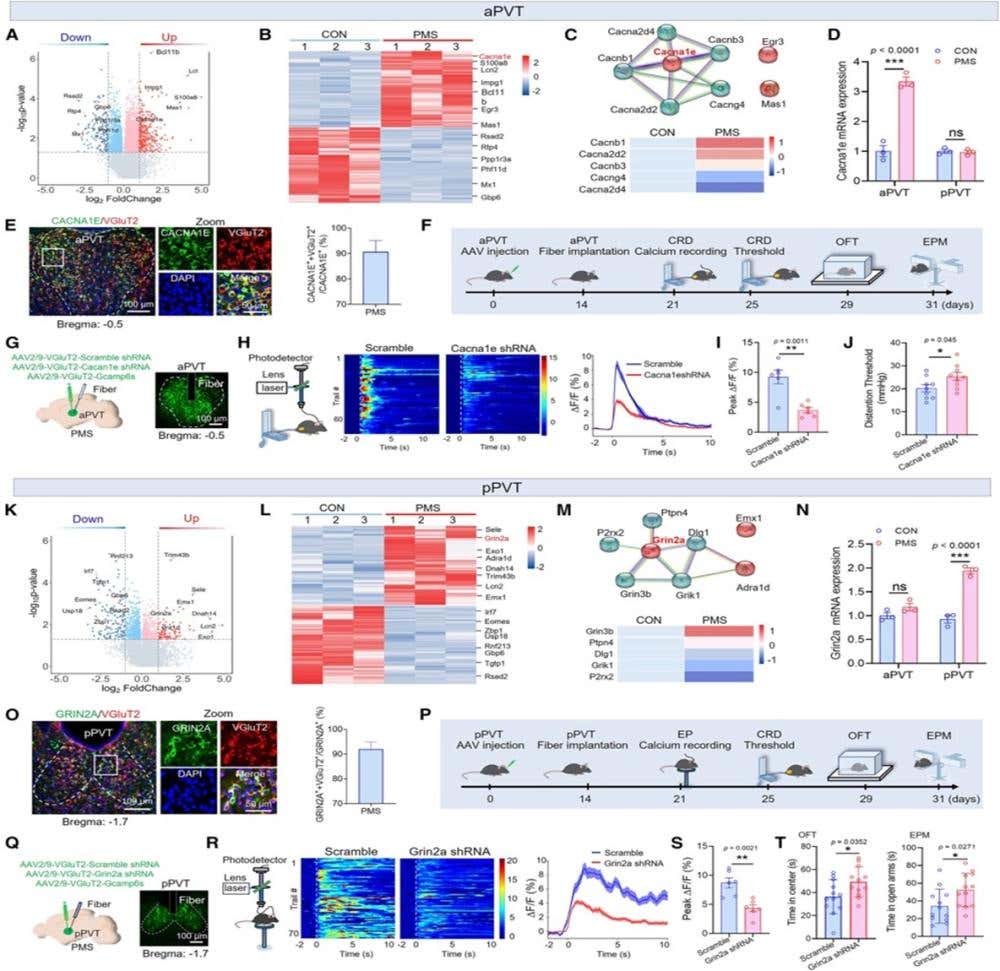
Figure 1. Cacna1e, selectively upregulated in aPVT, regulates visceral pain, whereas Grin2a, specifically upregulated in pPVT, mediates anxiety. (A–D) The screening process for Cacna1e in the aPVT. (A) Volcano plot showing DEGs in the aPVT of CON and PMS mice. (B) Heatmap of the top 30 most DEGs in the aPVT. (C) Protein correlation analysis plot (top), with a heatmap of gene expression differences closely associated with Cacna1e. (D) Statistical chart of Cacna1e expression changes in the aPVT and pPVT. (E) Representative images and statistical data showing co-labeling of CACNA1E (green) and VGluT2 (red) in the aPVT (n = 3 mice). (F–J) Knockdown of Cacna1e in aPVTGlu alleviates visceral pain in PMS mice. (F) Experimental flowchart for Cacna1e intervention. (G) Viral strategy and fluorescence images of Cacna1e knockdown. (H–J) Calcium activity heatmaps, curve plots, and average peak ΔF/F (n = 6 mice) of aPVTGlu during CRD stimulation in Scramble and Cacna1e knockdown groups, along with visceral pain thresholds (n = 9 mice) for both groups. (K–N) The screening process for Grin2a in the pPVT. (K) Volcano plot showing DEGs in the pPVT of CON and PMS mice. (L) Heatmap of the top 30 most DEGs in the pPVT. (M) Protein correlation analysis plot (top), with a heatmap of gene expression differences closely associated with Grin2a. (N) Statistical chart of Grin2a expression changes in the aPVT and pPVT. (O) Representative images and statistical data showing co-labeling of GRIN2A (green) and VGluT2 (red) in the pPVT (n = 3 mice). (P–T) Knockdown of Grin2a in pPVTGlu alleviates anxiety in PMS mice. (P) Experimental flowchart for Grin2a intervention. (Q) Viral strategy and fluorescence images of Grin2a knockdown. (R and S) Calcium activity heatmaps, curve plots, and average peak ΔF/F (n = 6 mice) of pPVTGlu during EP stress in Scramble and Grin2a knockdown groups. (T) Statistical data on the time spent in the center area of the open field and the open arms of the EPM for Scramble and Grin2a shRNA groups (n = 9 mice). Statistical analysis was performed using Student’s t test in (I), (J), (S), and (T), and two-way ANOVA followed by Bonferroni’s post hoc test in (D) and (N). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant.
Image and legend from Li et al. (2025). DOI: 10.1016/j.neuron.2025.04.019.
The Asymmetry
This is where the research delineates a genuine shift in how the brain sorts of pain from anxiety. The aPVT pain pathway projects primarily to the basolateral amygdala (BLA), which projects to GABAergic neurons in the central amygdala (CeA). The pPVT anxiety pathway skips the BLA and projects to the CeA GABAergic neurons directly. Here, we have two parallel routes converging on the same downstream node, as expected. But the routes aren't equivalent. Inhibit the aPVT and you reduce both pain and anxiety. Inhibit the pPVT and you only reduce anxiety – pain is unaffected. The aPVT-BLA-CeA circuit drives visceral pain and also feeds the anxiety machinery downstream; in contrast, the pPVT-CeA circuit modulates anxiety in isolation.
Li and colleagues directly tested this hypothesis. They found that selectively inhibiting just the BLA-to-CeA terminals of the aPVT-BLA circuit – sparing the BLA output that drives pain itself – leaves visceral pain thresholds unchanged but still relieves anxiety. The visceral pain signal is moving into the anxiety circuit through the CeA without needing to be experienced as pain at that point. Thus, pain becomes anxiety, but anxiety does not become pain.
For the ultimate confirmation, Li et al. found that inhibiting both circuits simultaneously relieves anxiety more than inhibiting either one alone – exactly what you'd expect if two non-redundant anxiety inputs converge on the CeA, and exactly what you wouldn't see if pain and anxiety were a closed feedback loop.
Limitations and Implications
The asymmetry unraveled here is the big story. In this model, the visceral pain circuit drives anxiety; the anxiety circuit does not drive pain. The implication for IBS anxiety comorbidity is clear: treat the visceral pain effectively and you may reduce a meaningful slice of the anxiety as a downstream effect but treat the anxiety alone and you should not expect the pain to be affected. The classic “vicious cycle” framing – pain and anxiety as a symmetrical closed loop – is probably wrong in at least one direction. The authors mention that their work offers “a theoretical foundation for personalized clinical treatment and targeted drug development.”
It’s also worth noting here that the molecular targets are interesting but need to be thought about in context. Cacna1e and Grin2a are knockdown validated here, in this circuit, in mice. Both encode broadly expressed channels with extensive roles elsewhere, and subregion-specific targeting in humans isn’t a near-term proposition.
What the data do establish, is that the PVT is not one nucleus doing one job. It's two subregions doing two jobs, with two different molecular substrates, two different downstream targets, and a one-way street between them. This indicates that in this system, visceral pain is upstream of anxiety – not its peer. That asymmetry was not visible when the PVT was treated as a single structure.


 Antibody")
 (extracellular) Antibody")


