
How oxytocin (OT) fits into a dedicated circuit, which calms your heart down after stress.
You’ve probably tried that trick where you take a long, slow breath when you’re stressed. And it actually works in a measurable, physiological way. When you take that breath, you heart rate does something interesting: it speeds up slightly when you breathe in and slows down when you breathe out, and that rhythm becomes more pronounced the calmer you get.
This is referred to as respiratory heart rate variability, (RespHRV). It’s bigger in healthy people, in athletes, in the young. It collapses in anxiety, depression, heart failure, and even people with autism spectrum disorder. But until now, nobody had pinned down exactly how your emotional state affects your heartbeat.
A paper published in Nature Neuroscience has a pretty solid answer. Your emotional state affects your heartbeat via a neural circuit in which oxytocin – the same molecule known for bonding, hugging, and being wildly over-explained in wellness content – is the signal that turns RespHRV up (1).
First, what is RespHRV?
Your heart doesn’t beat at a perfectly constant rate. Instead, it speeds up and slows down continuously, and the biggest driver of that variation is your breathing. During inhalation, your heart rate rises a little. During exhalation, it falls. The size of that swing is RespHRV, but the interesting part is what controls the size of that swing – because that’s where emotional state enters the picture.
When you’re calm, the swing is large. When you’re stressed, anxious, or ill, the swing shrinks. In some conditions it disappears entirely or even flips negative. Breathing techniques like pranayama and box breathing all amplify RespHRV by slowing the breath – the question has always been how.
The suspect: Oxytocin
Oxytocin (OT) is a neuropeptide produced in a region of the brain called the paraventricular nucleus (PVN), found deep in the hypothalamus. It’s been linked to social bonding, stress relief, and trust since the 1990s – often more enthusiastically than the evidence warranted. But one piece of evidence was always intriguing: intranasal OT administration in humans measurably increases RespHRV (2). Something connects OT to the heart rate swing, but nobody knew what, so the researchers decided to map it.
The Circuit, Step by Step
The heart’s moment-to-moment rhythm is controlled by the parasympathetic nervous system via the vagus nerve. A cluster of neurons in the brainstem called the nucleus ambiguus (nA) sends parasympathetic signals to the heart to slow it down. During inhalation, these neurons are inhibited – their brakes come off the heart, so rate rises. During exhalation, they’re active again, slowing things back down. RespHRV is essentially the depth of that pulsing inhibition.
That inhibition during inhalation comes from the brainstem’s respiratory pacemaker: the pre-Bötzinger complex (preBötC), a small cluster of neurons that generates the inspiratory rhythm. Every time you breathe in, the preBötC fires and some of these neurons simultaneously inhibit the cardiac neurons in the nA, lifting heart rate.
What the team found is that a specific subpopulation of preBötC neurons expresses the oxytocin receptor (OT-R). These aren’t the neurons generating the rhythm itself since inhibiting them doesn’t stop breathing. They consist of a modulatory layer within the preBötC, of approximately 700 neurons distributed bilaterally in mice, and they’re predominantly inhibitory (most are glycinergic meaning they release glycine, an inhibitory neurotransmitter). Interestingly, they connect directly to the cardiac neurons in the nA.
The fully assembled circuit comprises the following: hypothalamus (PVN) → OT → preBötCOT-R neurons → glycine → nA cardiac neurons → parasympathetic nerve → heart.
When OT arrives at those preBötCOT-R neurons, they become more excitable. During each inspiratory burst, they fire more strongly. The glycinergic inhibition they send to the cardiac neurons gets amplified. The swing in parasympathetic activity – and therefore in heart rate – gets larger, and RespHRV goes up.
How they proved It
In order to establish how the OT circuit works, the team used a thorough combination of approaches. Optogenetic analysis of the PVN OT fibers in the preBötC/nA region amplified RespHRV by 56% in freely moving mice. Injecting an OT-R blocker directly into the preBötC before stimulation almost completely abolished the effect, confirming it was specifically OT-mediated and not a side effect of the other neurotransmitters OT neurons also release.
To confirm that these preBötCOT-R neurons were responsible for the RespHRV response, the team needed to verify exactly which cells they were manipulating. When they delivered a bidirectional optogenetic tool (somBiPOLES) to OT-R-expressing cells in the preBötC, they used Alomone’s rabbit anti-oxytocin receptor antibody [Anti-Oxytocin Receptor Antibody (#AVR-013)] to immunolabel the tissue and confirm that the viral construct had landed in OT-R+ neurons (Figure 1). That antibody-based verification step mattered because it was confirmation that the neurons being switched on or off were genuinely the OT-R expressing population, and not just bystanders.
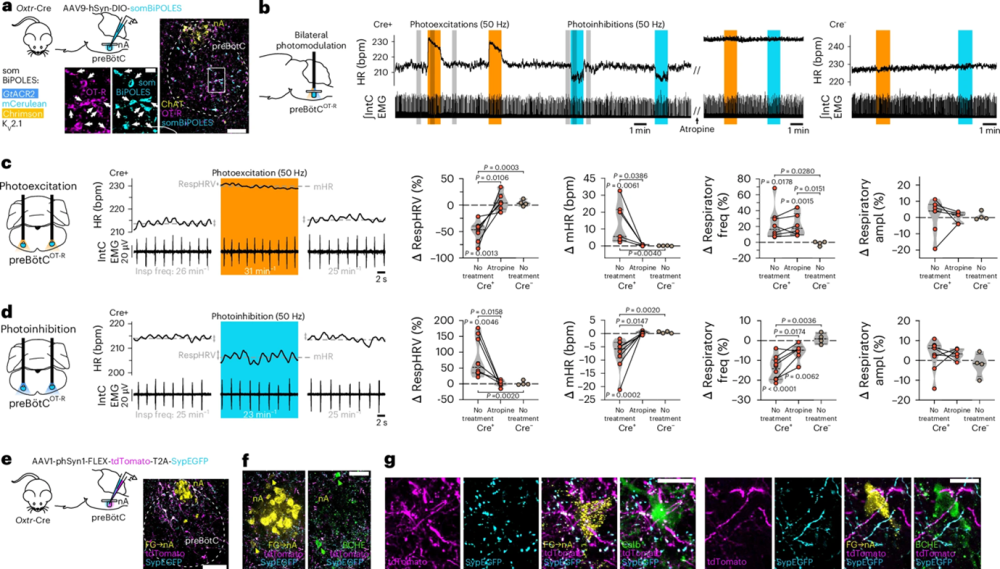
Figure 1. PreBötCOT-R neurons control respiratory heart rate variability (RespHRV) amplitude and project onto nA cardiac neurons. a, Viral-mediated expression of the somBiPOLES optogenetic construct in preBötCOT-R neurons of Oxtr-Cre mice (Extended Data Fig. 7i) (n = 10 mice). Viral injections in Cre− mice did not induce somBiPOLES expression (Extended Data Fig. 7h; n = 4 mice). An anti-Oxytocin Receptor (OT-R) antibody was used for immunohistochemical labeling. Scale bars: full image, 100 µm; inset, 20 µm. b, Effects of bilateral photomodulations of preBötCOT-R neurons in an anesthetized mouse (Cre+) on HR and inspiratory activity (IntC EMG), before and after intraperitoneal injection of the muscarinic receptor antagonist atropine (10 mg kg−1, 600 µl), which was compared to a control (Cre−) mouse. c,d, Expanded traces from the gray shaded areas in b, and individual data showing bidirectional cardiorespiratory effects of photoexcitation (c) vs. photoinhibition (d) of preBötCOT-R neurons (n = 8 Cre+ mice for photoexcitation and n = 10 Cre+ mice for photoinhibition without treatment; n = 6 Cre+ mice for photoexcitation or photoinhibition before vs. after atropine; n = 4 Cre− mice). Intra-group Δ changes (photoexcitation vs. pre-photoexcitation), were analyzed with a repeated measures one-way ANOVA with Tukey’s multiple comparison except for Cre− RespHRV photoinhibition, which was analyzed with a Friedman test with Dunn’s multiple comparison. Cre+ before and after atropine comparison, was analyzed with a two-sided paired t-test except for respiratory amplitude photoexcitation, which was analyzed with a Wilcoxon two-sided matched pairs signed-rank test. Inter-group comparison (Cre+ no treatment vs. Cre+ atropine vs. Cre− no treatment), was analyzed with a Kruskal–Wallis test with Dunn’s multiple comparison. Violin plots are represented in gray, dashed lines indicate the median and dotted lines represent the quartiles. Raw data are shown in Extended Data Fig. 7j,k. e, Viral-mediated expression of tdTomato and synaptophysin-enhanced GFP (SypEGFP) in preBötCOT-R neurons (n = 4 mice). Scale bar, 100 µm. f,g, PreBötCOT-R neurons project to (tdTomato+ fibers) and make putative presynaptic contacts with (SypEGFP+ puncta) nAcardiac neurons [FG+ butyrylcholinesterase+ (BCHE+) and FG+ calbindin+ (Calb+) neurons; Extended Data Fig. 7l; n = 4 mice]. Scale bars: in f, 50 µm; in g, 20 µm.
Figure taken from Buron et al. (2025). https://doi.org/10.1038/s41593-025-02074-2.
Exciting those neurons suppressed RespHRV. Inhibiting them amplified it. Blocking the parasympathetic nerve with atropine eliminated both cardiac effects, confirming the whole thing runs through the vagus. Sympathetic activity remained unchanged.
In brainstem slice recordings from newborn mice (where the preBötC keeps firing rhythmically even when removed from the body), adding the OT-R agonist [Thr4,Gly7]-OT (TGOT) amplified the inhibitory currents arriving at nA cardiac neurons during each inspiratory burst by over 50%. Blocking glycine receptors abolished those currents. The cellular mechanism was now clear: (1) OT depolarizes the OT-R-expressing, glycinergic preBötC neurons; (2) they fire harder during inspiration; and (3) the cardiac neurons downstream get a stronger inhibitory pulse. This results in a larger rebound during expiration.
But Does it Do Anything During Real Life?
This is where the paper becomes more than a circuit map or an experience that we’re all familiar with. The team chemogenetically silenced all OT neurons in freely moving mice – using a system referred to as designer receptors exclusively activated by designer drugs (DREADD) that lets you switch neurons off with a drug. Next, the mice were subjected to a restraint stress test. Stress lowered the RespHRV in all groups by about 50%.
But recovery was different. Mice with silenced OT neurons took twice as long to restore their RespHRV – 60 minutes instead of 30. Every other cardiovascular and respiratory parameter recovered on the same timeline in both groups. Thus, the RespHRV restoration specifically depended on OT neurons. The circuit activates when animals calm down after stress and that’s precisely the behavior this pathway was built for.
The Implications of OT-mediated Effects on RespHRV
Breathing techniques that amplify RespHRV have been practiced for centuries without anyone knowing why they worked at the level of neural hardware. This paper identified a parallel, non-voluntary system that does something similar, and that activates specifically during recovery from stress rather than during baseline calm.
The authors note one therapeutically relevant feature: the effect of OT on RespHRV occurs without meaningfully changing respiratory rate or depth. A pharmacological approach targeting this circuit might amplify RespHRV in conditions where it’s pathologically suppressed – heart failure, anxiety disorders, autism – without carrying respiratory side effects. That remains a hypothesis since this is derived from rodent data, not a clinical trial. But the mechanism is now specific enough to test.
Your nervous system has a dedicated circuit for calming your heart down after stress that runs on oxytocin. And it was there the whole time.

 (extracellular) Antibody")
 (extracellular) Antibody")