
Olfaction is often dismissed as a second-tier sense. It’s visceral, fleeting, hard to describe, and easy to ignore. But biologically, smell is one of the brain’s most direct and reactive inputs. In just milliseconds, odorants activate specialized cells in the nose, which send signals straight to the cortex without a thalamic relay. This bypass makes olfaction both primitive and intimate: tightly linked to memory, immune function, and stress response.
And smell is not static. It adapts to airflow, reflects inflammation, encodes long-term memories, and flags early signs of neurological disease. Four recent studies revealed how olfaction serves as a real-time reporter of cellular stress, neural plasticity, and systemic disruption – from immune breaches to mechanical load to memory consolidation.
When the Brain’s Defenses Slip, Smell is the First to Go
Using a rat model to better understand multiple sclerosis (MS), researchers observed something unexpected: long before animals developed motor symptoms, their ability to smell began to falter (1). Tests of odor detection and discrimination revealed early deficits – days before typical signs such as hindlimb weakness.
The reason wasn’t demyelination. Instead, the blood–brain barrier in the olfactory bulb briefly failed, allowing immune cells and serum proteins to infiltrate. This breach triggered glial activation and neuroinflammation, marked by a surge in adenosine A1 receptor (A1R) expression on microglia and astrocytes. Using our knockout-validated Anti-Adenosine A1 Receptor Antibody (#AAR-006), the researchers localized this upregulation specifically to the olfactory bulb (Figure 1).
By the time motor symptoms appeared, the blood-brain barrier had resealed. The olfactory change, it turns out, was a fleeting but meaningful early warning – suggesting that smell can serve as a sentinel for brain-directed immune activity.
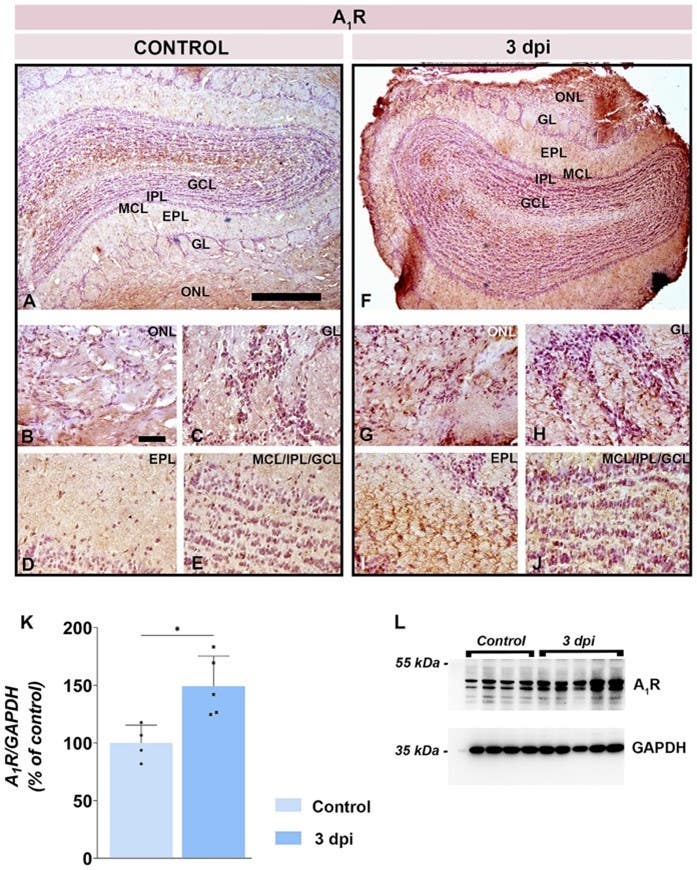
Figure 1. Early adenosine A₁ receptor (A1R) upregulation in the olfactory bulb during neuroinflammation. A-E) Representative immunohistochemical images from control rats showing baseline A₁R staining in the olfactory bulb (OB) layers: olfactory nerve layer (ONL), glomerular layer (GL), external plexiform layer (EPL), mitral cell layer (MCL), internal plexiform layer (IPL), and granule cell layer (GCL). F-J) Corresponding sections from rats at 3 days post-immunization (dpi) in the EAE model of MS, showed markedly increased A₁R immunoreactivity, most prominently along the pial surface and in the superficial OB layers (ONL and GL). K) A quantitative Western blot analysis demonstrated that A₁R protein levels in OB tissues are ~150% of control values at 3 dpi (p < 0.05). L) A representative Western blot of A₁R expression with a GAPDH loading control. Image adapted from Stekic et al., (2025).
Neurons Stay Young to Survive a Harsh Nose
In the olfactory system, some neurons don’t want to grow up – they hold onto immature traits as a survival strategy. New research showed that olfactory sensory neurons (OSNs) exposed to sustained environmental stress – specifically, elevated nasal airflow – retain juvenile molecular features to stay alive (2).
Using mice with genetically labeled mature OSNs, the researchers found that environmental stress triggered a stall in maturation. This block in maturation was marked by sustained nuclear expression of ATF5, a transcription factor known to regulate cell stress responses. In ATF5-deficient mice, this delay failed to occur, and mature OSNs instead died off in response to the same stress.
To confirm the presence and fate of mature OSNs, the researchers used our antibody directed against CNGA2 [Anti-CNGA2 Antibody (#APC-045)] to label canonical mature olfactory neurons. The CNGA2 signal dropped under stress in ATF5-deficient mice, consistent with cell loss rather than adaptive stalling (Figure 2).
The results suggested that delayed maturation acts as a protective adaptation, allowing neurons to stay flexible under mechanical or environmental strain. ATF5 is positioned as a central regulator of this plasticity.
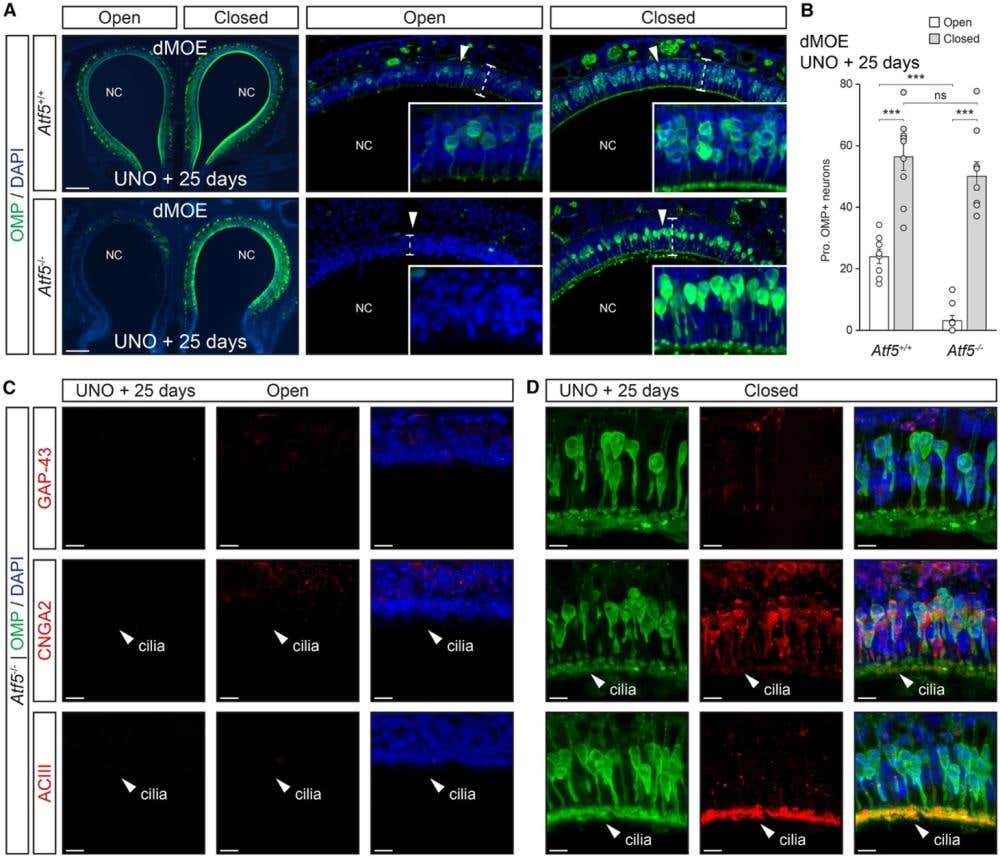
Figure 2. Olfactory neurons with juvenile-like traits are absent in ATF5 knock-out mice. A) Representative immunostaining for the olfactory marker protein (OMP, green) in the dorsal main olfactory epithelium (dMOE) of wild-type (Atf5⁺/⁺, white) and knock-out (Atf5⁻/⁻, gray) mice after 25 days of unilateral naris occlusion (UNO). In this model, one nostril is sealed to block airflow (“closed” side), while the other remains open to normal air exposure (“open” side). The white arrowheads indicate zoom-in regions, and dashed lines outline epithelial thickness. The nuclei were stained with DAPI. NC indicates the nasal cavity. Scale bars represent 200 μm. B) Quantification of the proportion of OMP-positive neurons (OMP⁺/DAPI⁺) in the dMOE for each genotype and condition shows that Atf5⁻/⁻ mice have significantly fewer mature olfactory neurons on the closed side compared to wild-type controls (two-tailed Student’s t-test or Mann-Whitney U-test, p < 0.001). The data are expressed as the mean ± SEM with aligned dot plots for ≥5 slices emerging from at least 3 different mice per genotype. C and D) Double immunostaining for OMP (green) and either GAP-43, CNGA2, or ACIII (red) illustrates the molecular differences between open (C) and closed (D) sides in Atf5⁻/⁻ mice. On the open side, CNGA2 and ACIII maintain their normal ciliary localization (white arrowheads), while this organization is lost on the closed side, indicating disrupted neuronal maturation and polarity. Scale bars represent 10 μm. Image adapted from Brechbühl et al., (2025).
The Cold Receptor Learns to Stay Quiet
Most of us will associate olfaction as a means to track pleasant smells, but it also senses irritants and temperature shifts. TRPM8, best known as the menthol and cold receptor, plays a role in detecting cool, chemical sensations in the nasal cavity. But rather than just fire and reset, the channel adapts and enters a desensitized state after sustained stimulation.
A new paper showed that TRPM8 inhibitors don’t simply block the channel in its open state (3). Instead, they bind selectively to its desensitized conformation, effectively locking it closed. Cryo-EM structures and electrophysiological assays – using our small molecule TRPM8 inhibitor, AMG2850 (#A-340) – showed that the inhibitor stabilizes the inactive state and prevents reactivation by new stimuli (Figure 3).
The results reframe how inhibition works: not by brute force, but by exploiting the channel’s own adaptation mechanism. This approach could prove useful in tuning down sensory hypersensitivity without wiping out baseline detection.
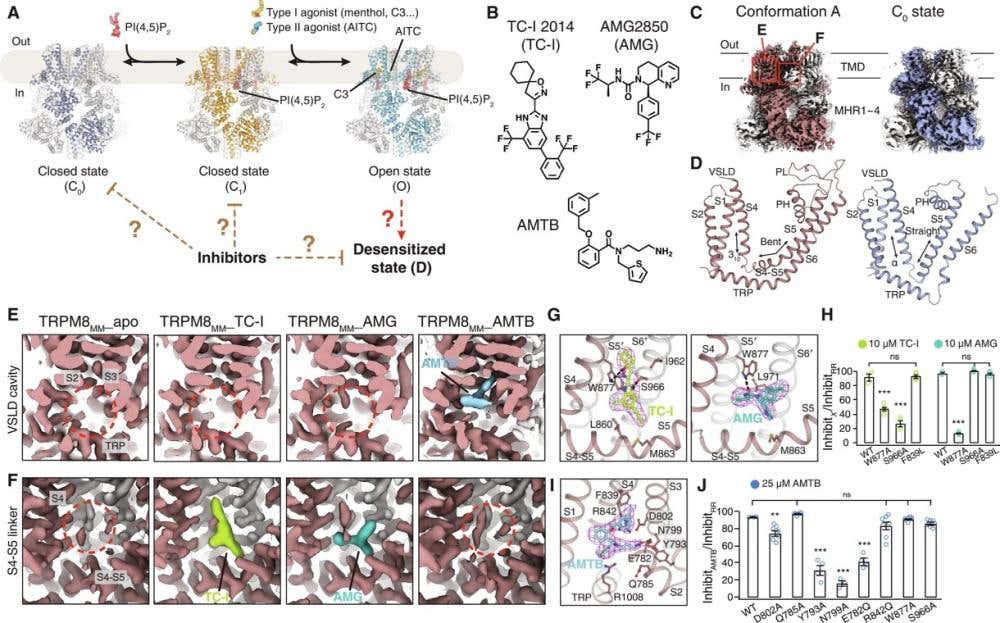
Figure 3. Cryo-EM structure determination of the TRPM8MM in complex with antagonists. A) Cartoon diagram illustrating the PIP₂- and cooling agonist–dependent gating pathway of TRPM8 channels. Published TRPM8MM structures in the C₀, C₁, and O states (PDB: 8E4P, 8E4N, and 8E4L, respectively) are used for reference. TRPM8MM is a stabilized mutant form of the mouse channel (M978L and M1000L) optimized for cryo-EM imaging. B) Chemical structures of the TRPM8 antagonists TC-I 2014, AMG2850, and AMTB. C) 3D reconstructions of the newly identified Conformation A (left, brown) and the previously known C₀ (closed) state (right, silver-gray). Neighboring protomers are shown in gray for clarity. D) Comparison of the transmembrane domains (TMDs) between Conformation A (left, brown) and the C₀ state (right, silver-gray). The S2–S3 linkers and the S3 helix were omitted to better highlight structural differences. E, F) Electron microscopy (EM) density maps showing the VSLD cavity (E) and the S4–S5 linker (F) from the Conformation A reconstruction in (C). TRPM8MM_apo represents the unbound channel. Densities corresponding to antagonists are colored lime for TC-I 2014, teal for AMG2850, and blue for AMTB. Red dashed circles indicate where antagonist density is absent. The maps are displayed at different visualization thresholds: 0.3 in (E) (showing broader, weaker signals) and 0.6 in (F) (highlighting only the strongest, most confident density regions). G, I) Binding sites and EM densities for TC-I 2014 [(G) left, lime sticks], AMG2850 [(G) right, teal sticks], and AMTB [(I) blue sticks]. The densities in magenta mesh are contoured at threshold levels of 0.3 for TC-I 2014, 0.34 for AMG2850, and 0.2 for AMTB. H, J) Summary of TRPM8 current inhibition by 10 μM TC-I 2014, 10 μM AMG2850 (H) or 25 μM AMTB (J), measured using two-electrode voltage clamp (TEVC) recordings in wild-type (WT) and TRPM8MM channels activated by 10–30 μM C3 (a potent synthetic TRPM8 agonist) at −60 mV. The inhibition was quantified as the percentage of current blocked by each antagonist relative to full inhibition by 50 μM Ruthenium Red (RR), a non-specific cation channel blocker. Individual oocytes are shown as open circles with the mean ± SEM (n = 3–8 oocytes). ns > 0.05; **P < 0.01; ***P < 0.001 by one-way ANOVA followed by Dunnett’s post hoc test. Image adapted from Yin et al., 2024.
The Nose Remembers: Plasticity in the Cortex Begins with NMDA Receptor Shifts
While the olfactory bulb handles initial signal processing, the cortex gives smell its meaning – linking it to memory and context. The following study zeroed in on how synapses involved in memory (engrams) adapt after learning. The research revealed how memory persistence in the cortex depends on a post-encoding switch in NMDA receptor subtypes at engram synapses: from GluN2B-containing NMDA receptors (linked to immature, highly plastic synapses) to GluN2A-containing receptors (associated with more mature, stabilized synapses) (4).
Shortly after learning, cortical engram neurons retain a strong GluN2B component. But over the next 30 days, GluN2B is gradually lost from the synapse, replaced by GluN2A. Using anti-GluN2A antibodies and our Anti-NMDAR2B (GluN2B) (extracellular) Antibody (#AGC-003), the team mapped this redistribution across time and cell type (Figure 4).
Preventing GluN2B exit from synapses early on disrupted memory consolidation. Conversely, stabilizing GluN2B at late stages preserved memory, suggesting that this receptor remains a gatekeeper for long-term plasticity. These effects occurred without changes in NMDA receptor amplitude, pointing to qualitative rather than quantitative shifts in synaptic signaling.
Though not olfactory-specific, the study sheds light on how odor-linked memories – often among the strongest we form – might be encoded and stabilized at a molecular level.
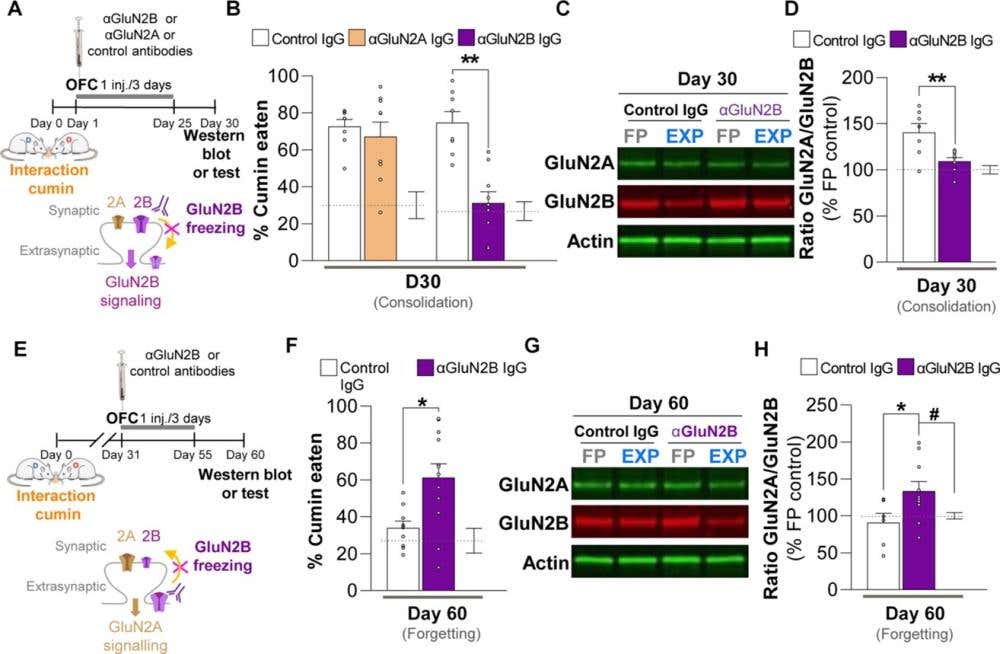
Figure 4. Modulating redistributions of GluN2B-containing NMDA receptors at cortical synapses determines the fate of remote memory. A) Rats received intra-orbitofrontal cortex (OFC) infusions of anti-GluN2B IgG, anti-GluN2A IgG, or control IgG during the early post-encoding period (Days 1–3), after learning to associate a food containing cumin with an aversive experience. B) Remote memory for this association was tested 30 days later by measuring how much cumin-flavored food the rats avoided (lower consumption = stronger memory); ** P = 0.0003. GluN2B blockade significantly impaired remote memory retrieval, whereas anti-GluN2A had no effect. C-D) Western blot (C) analysis of OFC tissue showed that learning normally increases the ratio of GluN2A- to GluN2B-containing NMDA receptors at synapses; this increase was abolished in rats treated with anti-GluN2B IgG (D); **P < 0.01, n = 8 to 9 rats per group. FP = Food-paired, EXP = Experimental. E) A parallel experiment examined the role of GluN2B signaling at a later time point – during the remote memory phase (Days 31–33). F) Blocking GluN2B at this stage weakened the remote memory when tested on Day 60, indicating a role for GluN2B in memory maintenance or forgetting processes; *P < 0.05, n = 10 to 14 rats per group. G-H) Western blot (G) results demonstrated corresponding changes in receptor subunit composition: GluN2B blockade altered the GluN2A/GluN2B ratio compared to controls (H). The Western blots in (C) and (G) were used for quantification of the respective GluN2A-/GluN2B-NMDAR ratios. Image adapted from Bessières et al. (2024).
The Nose Knows, and Then Some
Across four diverse contexts – autoimmune inflammation, mechanical stress adaptation, sensory channel inhibition, and cortical memory remodeling – olfaction proves to be more than a sensory gateway. It reflects how neurons respond to threat, how barriers break down, and how memories form and persist.
The olfactory system adapts to the world we live in; sometimes by rewiring its epithelium, sometimes by stalling development, and sometimes by tuning down its own excitability. And what starts in the nose often ends deep in the brain.
The nose, it turns out, is how the brain listens to the body.
Up Next: Hearing, Where Timing Becomes Everything
If smell shows how the brain senses stress, hearing reveals how it keeps time. In the next installment of the 5 Senses Series – Timing Is Everything: What Hearing Science Reveals About Precision, Protection, and Repair – we explore how the auditory system transforms microscopic mechanical deflections into millisecond-precise neural codes.



 (extracellular) Antibody")