And how TMEM132B, a single-pass transmembrane protein with no previously characterized function in neurons, revealed more than we expected.
Around 1.4% of the global population meet criteria for alcohol use disorder (AUD), a figure that translates to approximately three million deaths annually. We know the pharmacological targets of alcohol in the brain, but we still don’t fully understand how alcohol produces its characteristic effects well enough to consistently intervene therapeutically. A study published in Cell (1), identifies a molecular component that may account for a substantial part of that knowledge gap.
Alcohol and the Inhibitory Synapse
The central nervous system maintains excitatory-inhibitory balance through two primary ligand-gated systems: glutamate drives excitation and GABA drives inhibition. GABAA receptors (GABAARs) are the principal mediators of our brains’ fast inhibitory synaptic transmission. GABAARs are pentameric chloride channels, usually assembled from two α subunits, two β subunits, and one γ subunit, drawn from a pool of nineteen known subunit genes. This combinatorial diversity produces a large family of functionally distinct receptor subtypes distributed across brain regions and cellular compartments. Synaptic receptors (typically containing α1–3 and γ2) mediate phasic inhibition, while extrasynaptic receptors (typically containing α4–6 and δ) mediate tonic inhibition through persistent, low-level GABA signaling.
Alcohol acts on multiple ion channel targets. It inhibits NMDA-type glutamate receptors, dampening excitatory drive, and it potentiates glycine receptors. But its positive allosteric modulation of GABAARs – enhancing inhibitory chloride currents in response to GABA – is central to the anxiolytic, sedative, and hypnotic effects of alcohol. This is the same site of action shared by benzodiazepines, barbiturates, neurosteroids, and general anesthetics, which is why the behavioral profile of alcohol overlaps so much with that pharmacological class.
The problem is that the allosteric effect of alcohol on GABAARs has been difficult to attribute to GABAAR-specific subunit combinations. Alcohol appears to act across synaptic and extrasynaptic populations alike, and genetic deletion or mutation of individual GABAAR subunits in mice typically produces only narrow, subtype-specific changes in alcohol-related behaviors. Thus, an additional factor regulates the access of alcohol to the receptor at a level above subunit composition.
A Starting Point in the Alcoholic Brain
In the Cell paper, the researchers approached this problem by asking what changes at the membrane proteome level in AUD. They applied quantitative mass spectrometry to membrane proteins isolated from postmortem hippocampal tissue from six individuals with AUD and six matched controls, reasoning that altered expression of cell surface proteins (receptors, ion channels, and their modulatory partners) might point toward the molecular machinery that chronic alcohol consumption disrupts.
Among the proteins most consistently reduced in AUD samples was TMEM132B, a single-pass transmembrane protein with no previously characterized function in neurons. The reduction of TMEM132B in AUD samples was confirmed by Western blotting. And the result wasn’t specific to human postmortem tissue: mice given 15% v/v alcohol to drink for eight consecutive weeks showed reduced TMEM132B protein in the hippocampus, ventral tegmental area (VTA), and nucleus accumbens, with RT-qPCR confirming decreased transcript levels at all three sites. A luciferase reporter assay in mouse Neuro-2A (N2A) neuronal cells – which express endogenous TMEM132B – showed that alcohol directly suppresses TMEM132B promoter activity.
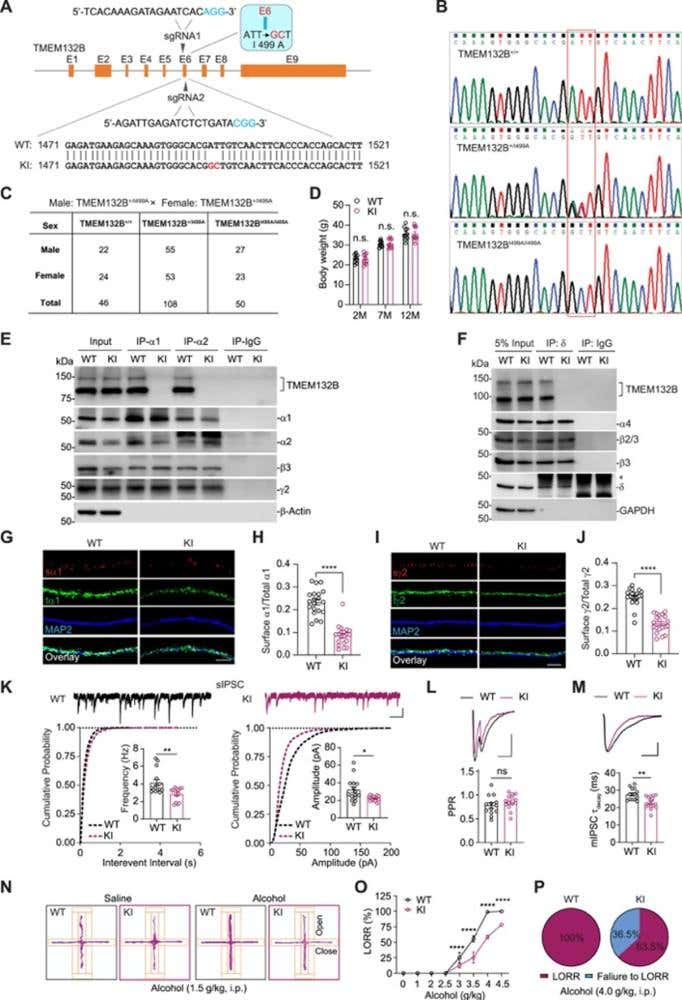
Figure 1. Generation and characterization of TMEM132BI499A knockin (KI) mice. (A) Strategy using two sgRNAs to generate the I499A point mutation in TMEM132B exon 6 (E6). The donor RNA was generated to convert the I499 to A, and the blue base pairs indicate the required protospacer-adjacent motifs (PAMs). The ATT code for I was converted to the GCT code. GC nucleotides are displayed in red. (B) The genotype sequencing samples showing wild-type (WT), heterozygous, and homozygous TMEM132BI499A mice. (C) The proportions of newborn mice were examined using Mendelian laws of heredity. Male: TMEM132B+/I499A × female: TMEM132B+/I499A. A total of 204 mice were analyzed. (D) The bodyweight of WT and TMEM132BI499A KI mice at different ages. n = 10 per group, two-way ANOVA with Bonferroni’s multiple comparisons test. (E) CoIP of TMEM132B with GABAAR α1 or α2 subunits in hippocampal lysates prepared from WT, but not TMEM132BI499A KI, mice. (F) CoIP of TMEM132B with GABAAR δ subunit in hippocampal lysates prepared from WT, but not TMEM132BI499A KI, mice. (G–J) Representative maximum intensity projection images showing the immunostaining of surface and total GABAAR α1 (G) and γ2 (I) subunits in cultured hippocampal neurons prepared from WT and TMEM132BI499A KI mice. Quantification of surface/total GABAAR α1 (H) and γ2 (J) subunits indicated that surface expression of GABAARs was reduced in TMEM132BI499A KI neurons. α1: n = 20 per group; γ2: n = 21 per group; t-test. (K) sIPSC recordings in CA1 pyramidal neurons in acute hippocampal slices prepared from WT and TMEM132BI499A KI mice showing both sIPSC frequency (left) and amplitude (right) were reduced in TMEM132BI499A KI neurons. WT: n = 15; KO: n =14; t-test. Scale bars, 500 ms, 20 pA. (L) Top: the paired-pulse ratio (PPR) of IPSC recordings in CA1 pyramidal neurons in acute hippocampal slices prepared from WT and TMEM132BI499A KI mice. Bottom: bar graph showing that the PPR was not significantly changed in TMEM132BI499A KI neurons. WT: n = 13; KI: n = 14; t-test. Scare bar, 25 pA, 20 ms. (M) Top: peak normalized sample traces of mIPSCs recorded from WT or TMEM132BI499A KI hippocampal CA1 neurons. Bottom: bar graph showing that the mIPSC decay time constant (mIPSC τ decay) was significantly decreased in hippocampal CA1 neurons from TMEM132BI499A KI mice. WT: n = 15; KI: n = 14; t-test. Scale bar, 25 pA, 20 ms. (N) Representative cumulative movement maps of WT and TMEM132BI499A KI mice in the elevated plus maze (EPM) after saline (left) or alcohol (right) injection (related to Figure 7E). (O) Dose-response relationship of alcohol-induced loss of righting reflex (LORR) in WT and TMEM132BI499A KI mice (WT and KI: 0–2.0 g/kg, WT: n = 12, KI: n = 13; 2.5 g/kg, WT: n = 12, KI: n = 14; 3 g/kg, WT: n = 16, KI: n = 14; 3.5 g/kg, WT: n = 17, KI: n = 15; 4 g/kg, WT: n = 15, KI: n = 14; 4.5 g/kg, WT: n = 14, KI: n = 16; two-way ANOVA with Bonferroni’s multiple comparisons test). (P) Pie chart showing that alcohol at the hypnotic dose (4 g/kg) produced LORR in 100% of WT mice (left, n = 51) but only in ∼64% of TMEM132BI499A KI mice (right, n = 52). Error bars indicate mean ± SEM (D, H, J, K, L, M, and O). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns, not significant.
Image taken from Wang et al. (2024). DOI: 10.1016/j.cell.2024.09.006.
TMEM132B protein synthesis and degradation rates in hippocampal tissue were not detectably altered by chronic alcohol exposure; therefore, the regulation is transcriptional.
TMEM132B is a GABAAR Auxiliary Subunit
Glutathione S-transferase (GST) pull-downs from mouse hippocampal lysates, followed by mass spectrometry, identified GABAAR subunits as TMEM132B-binding partners – a result consistent with a prior proteomic database entry that had placed TMEM132B in GABAAR complexes without a functional follow-up. Co-immunoprecipitation in HEK293T cells confirmed that TMEM132B associates with α1, α2, α3, α4, α5, and γ2 subunits – but not β2 nor β3 – spanning both synaptic and extrasynaptic receptor populations. In mouse hippocampal lysates, antibodies against α1 and α2 pulled down endogenous TMEM132B; the TMEM132B antibody reciprocally co-precipitated α1, α2, β2, β3, and γ2, but not GluA1 (AMPA receptor) nor glycine receptor subunits. Characterization of the interaction of TMEM132B with extrasynaptic α4- and δ-containing receptors used Alomone’s Anti-GABA(A) α4 Receptor (extracellular) Antibody (#AGA-008) and Anti-GABA(A) δ Receptor (extracellular) Antibody (#AGA-014). These studies revealed that α4 and δ-containing receptors co-immunoprecipitated with TMEM132B in hippocampal lysates – demonstrating that the interaction is present in native brain tissue. In cultured hippocampal neurons, endogenous TMEM132B colocalized with GABAergic synaptic markers – vesicular GABA transporter and gephyrin – but not with PSD-95 nor GluA1, confirming its inhibitory synaptic localization in situ.
The interaction between TMEM132B and GABA(A)R required a single residue: isoleucine 499 in the N-terminal bacterial IgG-like domain 1 (BIG1) of TMEM132B. Mutation of isoleucine to alanine (I499A) abolished the co-immunoprecipitation in heterologous cells and in hippocampal lysates from knockin (KI) mice.
Effects of TMEM132B on GABAAR Function
TMEM132B co-expression with α2β3γ2 receptors in HEK293T cells increased surface GABAAR abundance, enhanced GABA-evoked whole-cell current amplitude, and slowed receptor deactivation – the rate at which currents decay following agonist removal – without altering desensitization kinetics nor the EC50 for GABA (~18 µM, unchanged). Decelerated deactivation was consistent across α1β3γ2 and α4β2δ subtypes as well. In TMEM132B knockout (KO) neurons, the miniature inhibitory postsynaptic current (mIPSC) frequency and amplitude were both reduced in hippocampal CA1 pyramidal neurons (compared to wild-type littermates), and the mIPSC decay was faster – a composite picture of reduced surface receptor density and altered current kinetics that aligns with the heterologous cell data.
A Specific Amplifier of the Allosteric Action of Alcohol
The most pharmacologically consequential finding was that TMEM132B mediated the selective enhancement of alcohol sensitivity. Co-expression of TMEM132B with α1β2γ2, α2β3γ2, α3β3γ2, or α4β2δ receptors each produced greater alcohol-induced potentiation of GABA-evoked currents than receptors expressed without TMEM132B. Dose-response analysis showed that TMEM132B increased the efficacy of alcohol – to the maximal degree of potentiation achievable – without shifting the EC50 (approximately 54 mM for α2β3γ2, unchanged with TMEM132B). None of the other GABAAR-associated proteins tested – Shisa7, gephyrin, LHFPL4, and neuroligin-2 – produced this effect on the same receptor subtype. In TMEM132B KO hippocampal neurons, alcohol-induced potentiation of GABA-evoked currents was strongly diminished; re-expression of wild-type TMEM132B rescued it fully, while TMEM132B(I499A) did not. Diazepam-induced potentiation was unaffected by TMEM132B KO, indicating specificity for the alcohol modulatory site rather than a general change in allosteric accessibility.
From Molecule to Behavior
TMEM132B KO mice were normal across a broad behavioral battery of tests including open field, rotarod, forced swim, marble burying, sucrose and salt preference, as well as food intake. But their responses to alcohol were substantially altered. At an anxiolytic dose of 1.5 g/kg (i.p.), alcohol increased open arm entries and open arm dwell time in the elevated plus maze in wild-type animals; it did not do so in TMEM132B KO mice. At sedative doses, 100% of wild-type mice showed loss of righting reflex (LORR) at 4 g/kg, compared to 62% of TMEM132B KO mice. Moreover, the LORR latency was prolonged, and its duration markedly shortened in those TMEM132B KO mice that did respond.
In TMEM132B KO mice, voluntary alcohol consumption moved in the opposite direction. The KO mice drank more at every concentration in a two-bottle choice test, and maintained elevated intake across pre-binge, binge, and post-binge phases of a compulsive drinking paradigm. This compulsive drinking behavior occurred even in the presence of quinine, a natural alkaloid used to model the persistence of drinking despite negative consequences. TMEM132B(I499A) KI mice replicated every phenotype of TMEM132B KO mice. Since the TMEM132B(I499A) mutation disrupts the TMEM132B-GABAAR protein-protein interaction without removing TMEM132B expression, the behavioral effects are attributable to the loss of this specific molecular interface.
The Implications
The pattern across TMEM132B KO and KI animals was internally consistent: the reduced TMEM132B-GABAAR interaction diminished alcohol-mediated potentiation of inhibitory currents, which attenuated the anxiolytic and sedative effects that normally accompany drinking. This, in turn drives increased consumption of alcohol – an attempt, at the circuit level, to recover a diminished pharmacological signal. Whether reduced TMEM132B expression in AUD brains reflects cause, consequence, or both remains an open question. What the data established is that the TMEM132B-GABAAR complex is a functional amplifier of the effects of alcohol on the inhibitory system, and that its disruption is sufficient to substantially alter the pharmacological and motivational consequences of drinking in vivo.


 α2 Receptor Antibody")
 γ2 Receptor (extracellular) Antibody")
 α4 Receptor (extracellular) Antibody")
 δ Receptor (extracellular) Antibody")