Adrenoceptors are seven transmembrane proteins belonging to the superfamily of G-protein coupled receptors (GPCRs). They respond to the endogenous ligands adrenaline and noradrenaline by inducing a cascade of physiological effects. These receptors have a central role in blood pressure regulation and smooth muscle relaxation and heart contraction. After Rhodopsin, they have become the most studied GPCR to date and are targets for the continuous development of agonists/antagonists. Alomone Labs offers an exhaustive list of antibodies related to adrenoceptors, enabling the never ending research done on adrenergic signaling. The wealth of information on these receptors is truly overwhelming; hence this very short review really glances over some of the important aspects of adrenergic signaling and its significance.
Introduction
Adrenaline and noradrenaline are two catecholamines (a monoamine) synthesized from tyrosine in the adrenal glands and secreted throughout the body. These catecholamines induce the “fight or flight” response occurring under stressful situations by exerting a cascade of physiological pathways via adrenergic receptors. Adrenergic receptors (or adrenoceptors) were originally categorized into α and β types. Following extensive pharmacological analysis, mainly consisting in radioligand binding studies, molecular analysis as well as their signaling properties, they are now divided into three major groups; α1-, α2– and β-adrenoceptors which are further divided.
This group of receptors belongs to the superfamily of G-protein coupled receptors (GPCRs) and therefore includes the classical features of GPCRs, namely a seven transmembrane spanning domain and the ability to couple a variety of G-proteins which ultimately determines the cellular output. In addition, adrenoceptors also undergo desensitization in order to regulate/terminate their signaling, a critical point which can be the source of many diseases. As will be elaborated below, adrenergic receptors are best known for their role in blood pressure regulation and cardiac functions as many agonists/antagonists have been and are being developed for the treatment of adrenoceptor signaling malfunctions.
α1-Adrenoceptors
Three subtypes of α1 receptors have been described and identified to date: α1A-, α1B– and α1D-adrenoceptors. All three receptors are known to couple Gq/11 and activate phospholipase C (PLC) which eventually leads to an increase in intracellular Ca2+. However, in the rat heart, α1-adrenoceptors have been seen to couple Gs, thereby leading to an increase in intracellular cAMP levels15. Many GPCRs can form homo and heterodimers and/or oligomers27. It also seems to be the case for α1D-adrenoceptor, which is the only receptor belonging to this family, that when heterologously expressed, is found mostly in intracellular compartments, and largely nonfunctional7,35. However, coexpression of α1D– with β2-adrenergic receptor increases its cell surface expression suggesting that α1D function and expression may involve heterodimerization35. The α1-adrenoceptors are responsible for mediating smooth muscle contraction. In the vascular system they are mainly responsible for controlling blood pressure10. In fact, hemorrhage, which decreases blood pressure, activates the baroreceptor reflex, adrenaline release and concomitantly vasoconstriction via α1-adrenergic receptors. In general, antagonists of α1-adrenoceptors lower blood pressure in hypertension (although not commonly used). Prazosin, a specific α1-adrenergic receptor blocker is administered to patients suffering from high blood pressure and is therefore quite safe as it does not affect cardiac functions. In the history of the adrenergic receptor classification, prazosin sensitivity was used as a classification prerequisite; α1H and α1L for high and low sensitivity, respectively. It became apparent that the α1H matches the sensitivity of α1A-, α1B-, and α1D-adrenoceptors10. The low prazosin affinity receptor, α1L-adrenoceptor, has been functionally detected in different tissues34. However, no gene has been detected to date. It is believed that α1L-adrenoceptor is a conformational state or a functional phenotype of the α1A-adrenoceptor13,26,34. Convincing evidence that this is in fact the case comes from a study in which α1A-adrenoceptor knockout mice do not display the α1L-adrenoceptor phenotype (the phenotype is apparent only when α1A-adrenoceptor is present)17,28. Agonists of these receptors are used for treating hypotension. These agonists reinforce the vasoconstrictive effects by a depolarizationmediated Ca2+ entry through L-type or T-type Ca2+ channels, or by releasing Ca2+ from intracellular stores8.
In the visual system, α1-adrenoceptors are responsible for contracting the dilator papillae, thereby evoking dilation of the pupil and increasing the amount of light reaching the retina. Patients suffering from intraocular pressure can be treated by the administration of α1-agonists, which may act by decreasing the blood flow in the area10.
Additional roles for α1-adrenoceptors include the induction of sleep, mediated by post-synaptic α1-adrenoceptors (and α2) in noradrenergic afferents to the preoptic area22, constriction in the gastrointestinal tract, salivary secretion, bronchoconstriction and a predominant role in the genitourinary system, including prostate proper function, ejaculation and bladder contraction (which may involve α1D-adrenoceptor)9,10.

Immunohistochemical staining of rat cerebellum using Anti-α1A-Adrenergic Receptor (extracellular) Antibody (#AAR-015), (1:100). A. α1A-adrenoceptor (green) appears in fibers of Bergmann glia (triangle points at an example). B. S100b (red), a marker of Bergmann glia, is stained in the same section. C. Merge of the images demonstrates expression of α1A-adrenoceptor in fibers of Bergmann glia. α1A-adrenoceptor also is expressed in cells in the molecular layer that are s100b negative (arrow points at an example). DAPI is used as the counterstain (blue).
Experimental procedure and figure processed at Alomone Labs.
α2-Adrenoceptors
Three α2-adrenoceptor subtypes have been identified to date, and well characterized in different mammalian systems: α2A-, α2B-, and α2C-adrenoceptors. α2D-adrenoceptor, a fourth receptor was originally identified in rat and mouse. Today, it is largely accepted that this receptor is in fact α2A-adrenoceptor displaying a slightly different pharmacological profile in these two organisms23,29. Nevertheless, it seems that the complexity of this adrenergic receptor category is indeed complex as some organisms express multiple α2-adrenoceptors; zebrafish and pufferfish express five and up to eight receptors although some may be gene duplicates6. All three receptors couple Gi/o thereby decreasing the activity of adenylyl cyclase. However, α2-adrenoceptors could also couple Gs and consequently increase intracellular cAMP levels19. Interestingly, it was suggested that at low agonist concentrations, α2-adrenoceptors decrease cAMP levels, whereas at high agonist concentrations, increased cAMP levels are observed11,21. α2A-, α2B– and α2C-adrenoceptors are widely distributed in the central nervous system and the periphery. The brain and its various regions express these receptors. In the periphery, the various subtypes can be found in platelets5, kidney3, gastric mucosa18, aorta, spleen, heart and liver3. This category of receptors participates in the proper physiological function of the cardiovascular system and the central nervous systems. Physiological roles attributed to these receptors include motor coordination, by α2-adrenoceptors expressed in the cortex20.
Not surprisingly, α2-adrenoceptors have been characterized through pharmacological studies, but since specific pharmacological tools are not available, the specific function of each subtype remains to be deciphered19. Nevertheless, α2A-adrenoceptors seem to mediate most of the classical functions of α2-adrenoceptors such as the antihypertensive and bradycardic effects observed by the actions of α2-agonists on the α2A-adrenoceptor1. α2A-adrenoceptor exhibits a dominant role in regulating the cardiovascular system, demonstrated by knockout mice studies. These mice demonstrate high blood pressure, increased heart rate and a higher probability to develop heart failure4. α2B-adrenoceptor is mostly post-synaptic as opposed to its α2A– and α2C-adrenoceptor counterparts. Also, α2B-adrenoceptor mediates the contractions of rat pregnant uterus, whereas α2A– and α2C-adrenoceptors inhibit these contractions in late pregnancy16. α2C-adrenoceptor is mainly localized to the CNS and may counteract α2-agonist sedative effects in mice31. This receptor subtype may also have beneficial effects in various psychiatric disorders including stressdependent depression, schizophrenia, as well as drug withdrawal. α2-adrenoceptor agonists are also being potentially used in the treatment of attention-deficit hyperactive disorder (ADHD)33.
While the α2A-adrenoceptor is the most abundant and is the most dominant pertaining to cellular functions, the α2B– and α2C-adrenoceptor subtypes can either contribute or counteract α2A-adrenoceptor dependent processes in addition to their own independent roles.
β-adrenoceptors
β-adrenoceptors have been classified into β1, β2 and β3 subtypes. Activation of β1-adrenoceptor leads to its coupling to Gs and subsequently to the increase in intracellular cAMP levels. β2– and β3-adrenoceptors couple both Gs and Gi rendering the β2-adrenoceptor signaling in cardiac cells more complicated36. Although all three receptors are expressed in the heart, and share expression in various tissues, β1-adrenoceptor is mostly expressed in the heart and regulates key events in the kidney. β2-adrenoceptor is mainly found in the airway smooth muscle (i.e. the respiratory tract) while β3-adrenoceptor is largely distributed in adipose tissue where it stimulates the mobilization of lipids from white fat cells and increases thermogenesis in brown fat cells2,14. The central role and involvement of β1-adrenoceptor in proper heart function is demonstrated by the fact that patients suffering from heart failure exhibit a decrease in β1-adrenoceptor cell surface expression which is partly due to the internalization of the receptor as well as its desensitization25.
Evidence regarding mutations and coding single nucleotide polymorphisms (cSNPs) is growing concerning their effect and incidence in genes encoding β-adrenoceptors. The polymorphisms inadvertently affect coupling and desensitization rates of the receptor. cSNPs have been observed for β1-adrenoceptors which affect the risk of cardiovascular disease as well as the response to treatment (namely antagonists)32. A missense mutation in the β2-adrenoceptor gene is strongly associated with obesity2. Also, a polymorphic form of the β3-adrenoceptor is also associated with obesity as well as diabetes (reviewed in ref. 2).
Activation of β-adrenoceptors activates pathways that eventually lead to receptor desensitization. The mechanisms engaged in turning off the signaling response include phosphorylation of the receptors. Desensitization of β-adrenergic receptors could be homologous, where solely activated receptors are phosphorylated; this requires GPCR kinase 2 and 3 (GRK2, GRK3). Heterologous desensitization is non-specific and non-activated receptors are also phosphorylated; this phosphorylation requires the activity of PKA and occurs rapidly following activation of the receptor. The phosphorylation of the β-adrenergic receptors eventually leads to the uncoupling of the receptor from the G-protein and its internalization via clathrin-coated pits where it is dephosphorylated and either recycled back to the cell surface or degraded24.
β-adrenoceptor agonists (β-agonists) were, for the longest time, administered to patients suffering from asthma in order to induce bronchodilation. At some point it became clear that some β-agonists also have the ability to increase muscle mass and decrease body fat12. Although this characteristic of β-agonists can be practical in the treatment of disorders like muscular dystrophy, β-agonists have also become abusive drugs in competitive bodybuilders and athletes wanting to improve their performance30.
The interest and research in adrenoceptors is still intensive after so many years. Alomone Labs offers antibodies to Adrenoceptors: Anti-α1A-Adrenergic Receptor (extracellular) Antibody (#AAR-015), Anti-α1B-Adrenergic Receptor (extracellular) Antibody (#AAR-018), the labeled version, Anti-α1B-Adrenergic Receptor (extracellular)-ATTO Fluor-488 Antibody (#AAR-018-AG), Anti-α1D-Adrenergic Receptor (extracellular) Antibody (#AAR-019), Anti-α2B-Adrenergic Receptor (extracellular) Antibody (#AAR-021), Anti-α2C-Adrenergic Receptor (extracellular) Antibody (#AAR-022), Anti-β2-Adrenergic Receptor (extracellular) Antibody (#AAR-016) and Anti-β3-Adrenergic Receptor (extracellular) Antibody (#AAR-017). Alomone Labs adrenergic receptor antibodies enable the detection of these receptors using various applications such as western blot, immunohistochemistry, immunocytochemistry, direct and indirect flow cytometry (see the adjoined figures).
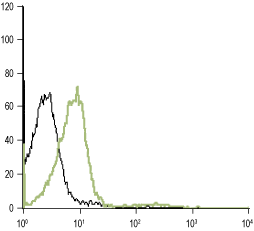
___ Unstained cells.
___ Cells + Anti-α1D-Adrenergic Receptor (extracellular) Antibody (#AAR-019), (5-10 µg antibody/0.5×106 cells).
Experimental procedure and figure processed at Alomone Labs.
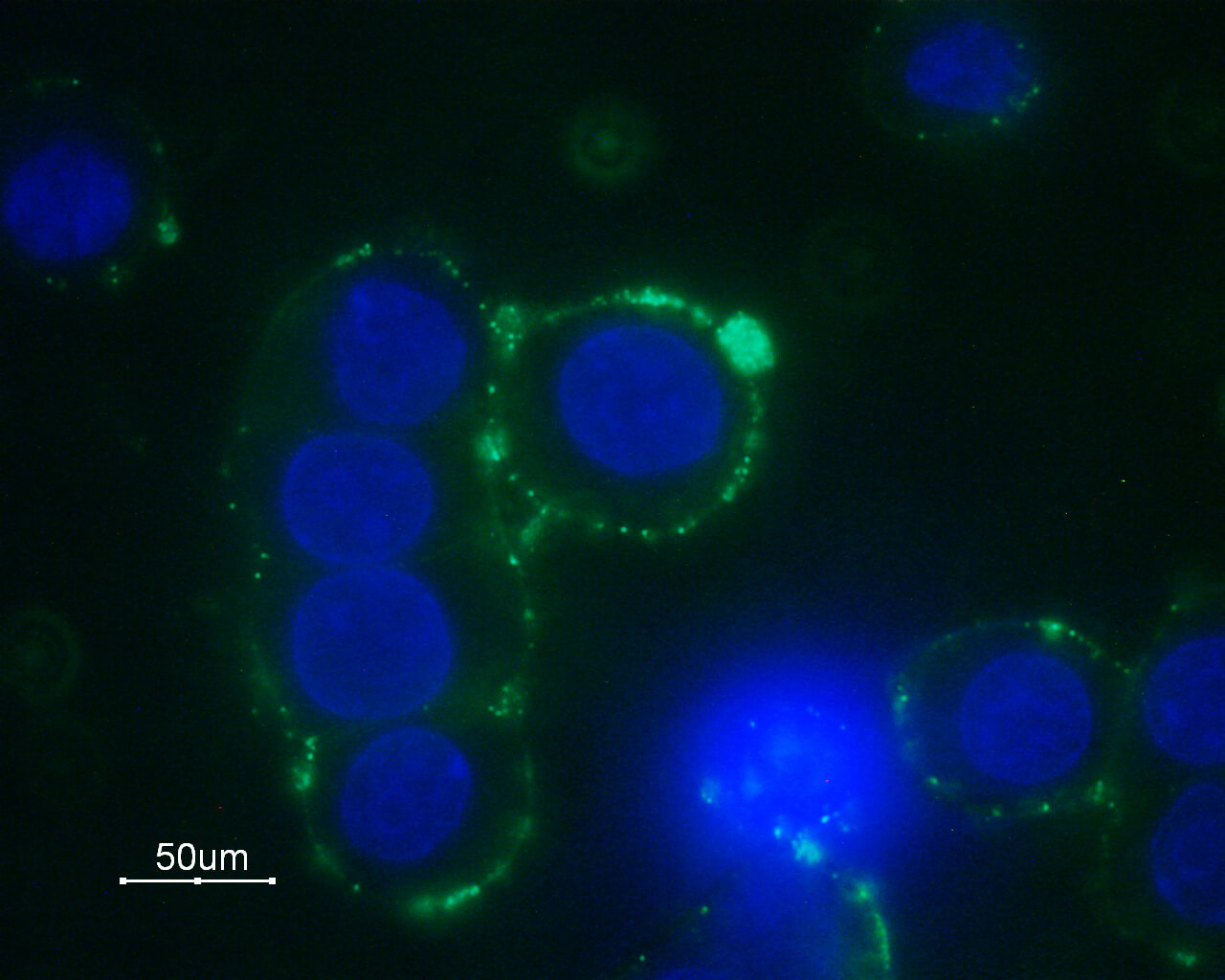
Immunocytochemical staining of GH3 cells with Anti-α1B-Adrenergic Receptor (extracellular) Antibody (#AAR-018), followed by goat anti-rabbit-AlexaFluor-488 secondary antibody (green). Nuclear staining of cells using the DNA dye Hoechst 33342 (blue).
Experimental procedure and figure processed at Alomone Labs.

Immunohistochemical staining of rat kidney paraffin embedded sections using Anti-α2C-Adrenergic Receptor (extracellular) Antibody (#AAR-022), (1:100). α2C-Adrenoceptor is expressed in both proximal (PT) and distal (DT) renal tubules and in the collecting ducts (CD). Hematoxilin is used as the counterstain.
Experimental procedure and figure processed at Alomone Labs.
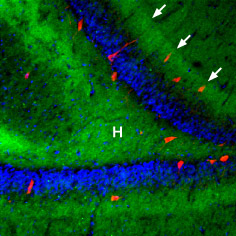
Immunohistochemical staining of rat hippocampal dentate gyrus using Anti-β3-Adrenergic Receptor Antibody (#AAR-017), (1:100), (green). β3-Adrenoceptor is strongly expressed in the hilus (H) and in the outer molecular layer (arrows). The distribution overlaps the entire layers, but is not restricted to nerve cells (stained red with mouse anti-Parvalbumin). DAPI is used as the counterstain (blue).
Experimental procedure and figure processed at Alomone Labs.
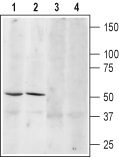
1, 3. Anti-β3-Adrenergic Receptor (extracellular) Antibody (#AAR-017), (1:200).2, 4. Anti-β3-Adrenergic Receptor (extracellular) Antibody, preincubated with the control peptide antigen.
Experimental procedure and figure processed at Alomone Labs.
References
- Altman, J.D. et al. (1999) Mol. Pharmacol. 56, 154.
- Arner, P. and Hoffstedt, J. (1999) J. Intern. Met. 245, 667.
- Blaxall, H.S. et al. (1994) Receptor 4, 191.
- Brede, M. et al. (2002) Circulation 106, 2491.
- Bylund, D.B. (1988) Trends Pharmacol. Sci. 9, 356.
- Bylund, D.B. (2005) Br. J. Pharmacol. 144, 159.
- Chalothorn D. et al. (2002) Mol. Pharmacol. 61, 1008.
- Chen, X.L. and Rembold, C.M. (1995) Am. J. Physiol. 268, H74.
- Chen, Q. et al. (2005) J. Urol. 174, 370.
- Docherty, J.R. (2010) Cell. Mol. Life Sci. 67, 405.
- Eason, M.G. et al. (1992) J. Biol. Chem. 267, 15795.
- Emery, P.W. et al. (1984) Biosci. Rep. 4, 83.
- Ford, A.P. et al. (1997) Br. J. Pharmacol. 121, 1127.
- Frielle, T. et al. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 9494.
- Gallego, M. et al. (2005) Am. J. Physiol. 288, C577.
- Gaspar, R. et al. (2007) Neurochem. Int. 51, 311.
- Gray, K. et al. (2008) Br. J. Pharmacol. 155, 103.
- Gyres, K. et al. (2007) Neurochem. Int. 51, 289.
- Gyres, K. et al. (2009) Neurochem. Int. 55, 447.
- Hirono, M. et al. (2008) Neuroscience 156, 143.
- Jasper, J.R. et al. (1998) Biochem. Pharmacol. 55, 1035.
- Kumar, V.M. et al. (2007) Neurochem. Int. 50, 783.
- Link, R.E. et al. (1992) Mol. Pharmacol. 42, 16.
- Lynch, G.S. and Ryall, J.G. (2008) Physiol. Rev. 88, 729.
- Marian, A.J. (2006) J. Mol. Cell. 41, 11.
- Marti, D. et al. (2005) Am. J. Physiol. 289, H1923.
- Minneman, K.P. (2007) Biochem. Pharmacol. 73, 1043.
- Muramatsu, I. et al. (2008) Br. J. Pharmacol. 155, 1224.
- O’Rourke, M.F. et al. (1994) J. Pharmacol. Exp. Ther. 271, 735.
- Prather, I.D. et al. (1995) Med. Sci. Sports Exercise 27, 1118.
- Puolivali, J. et al. (2002) Neuropharmacology 43, 1305.
- Sandilands, A.J. and O’Shaughnessy, K.M. (2005) Br. J. Clin. Pharmacol. 60, 235.
- Scahill, L. (2009) CNS Drugs 23, 43.
- Segura, V. et al. (2010) Eur. J. Pharmacol. 641, 41.
- Uberti, M.A. et al. (2005) J. Pharmacol. Exp. Ther. 313, 16.
- Zhu, W. et al. (2005) Circ. Res. 97, 507.