Introduction
Modulation of ion channel activity is a fundamental mechanism in many tissues. Ion channels, like many other proteins are targets for several intracellular signaling pathways, including protein phosphorylation and dephosphorylation. These processes can modify channel activity and dramatically alter the electrophysiological properties of both excitable and nonexcitable cells1-5. The amino acid sequences of ion channels suggest that many contain a wide variety of possible phosphorlyation sites. Phosphorylation in general and Tyrosine Phosphorylation (TP) in particular were shown to participate in the control of ion channel activity. Initial evidence showing that ion channels may be regulated by TP came largely from pharmacological studies using soluble inhibitors for Protein Tyrosine Kinases (PTKs) such as Genistein, K252a/b, tyrphostins, and lavendustin or by phosphotyrosine phosphatase inhibitors. Electrophysiology and molecular biology studies subsequently provided insight into the mechanism of TP regulated ion channel activity2-5. In this mini review, we describe some modulation pathways that affect different ion channels. This highlights the contribution of TP in ion channel regulation and the importance of their physiological modulation in many systems.
Cyclic Nucleotide-Gated (CNG) Channels
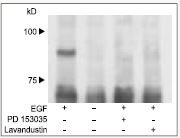
CNG cation channels play a critical role in many cellular processes especially in photoreceptors by transducing a decrease in cGMP into hyperpolarization during the light response in vertebrates6-8. The role of TP in regulating the CNG channel was demonstrated when orthovandate slowed the progressive sensitivity enhanced by Insulin Like Growth Factor-1 (IGF-1)9,10. The molecular mechanism of IGF -1 modulation involves changes in TP of CNGA1 and CNGB1 subunits. Moreover, TP was suppressed or reversed by application of ATP but not by non-hydrolyzable ATP analog. The effect was reduced but not entirely abolished by the mutation in Tyrosine 498F of the β–strand cAMP binding site. This is consistant with the idea that this residue is phosphorylated by PTK11-14. This is the regulating site of the Ca2+/Calmodulin (CaM) binding to the C terminus of the CNGB1 subunit. Phosphorylation at this site, triggered uncoupling of Ca2+/CaM from the CNGB1, thereby curtailing Ca2+/CaM inhibition15. This mechanism demonstrates that modulation of CNG channels by TP may regulate the sensitivity of cones and rods to light. The Tyrosine 59 of CNGA2 is a potential residue for TP by EGF receptor (EGFR) Tyrosine Kinase activity. In Fig 1, immunoprecipitation with Anti-CNGA2 antibody (#APC-045) was demonstrated by a phosphorylation of CNGA2 by EGFR, which was detected by phosphotyrosin specific antibody. Tyrphostin PD 153035 (#P-360) (a specific EGFR PTK inhibitor) and Lavandustin prevented the phosphorylation.
L-Type Ca2+ Channels
The L-type Ca2+ channel is the primary voltage-dependent Ca2+-influx pathway in many excitable and other secretory cells. Its pore-forming subunit, Cav1.X, contains NH2– and COOH-terminal cytosolic domains that are potential targets for protein phosphorylation. The activity of L-type Ca2+ channels can be regulated by different types of kinases, such as protein kinase A (PKA) and protein kinase C (PKC)16,17. Both are serine threonine kinases that phosphorylate the Cav1.2 and its auxiliary β subunits18,19. An emerging body of evidence suggests that these Ca2+ channels are also regulated by phosphorylation on tyrosine residues5.
Ca2+ channel EGF-complex was isolated from solubilized epithelial cell membranes of gastric mucosa. Furthermore, EGF enhanced the Ca2+ uptake by 48% in a reconstituted complex20. Receptor single cell microflurometry and whole-cell patch-clamp techniques, demonstrated that the activity of Ca2+ channels can be influenced by the interplay of PTK and PTP activity. A rise in [Ca2+]i elicited by the specific L-type Ca2+ channel activator (±)Bay K8644 (#B-350) or by high K+ concentrations, was reduced by PTK inhibitor Genistein (#G-300) and lavandustin. On the other hand, the PTP blocker vanadate, enhanced the rise of [Ca2+]i stimulated by (±)Bay K8644. It seems that PTK activation caused an increase, whereas PTP activation appeared to exert an inhibitory role on these ion channels. Cataldi, M. et al. demonstrated that TP modulates the activity of L-type channels in the resting state, as well as in the activated state5.
At least two isoforms of the L-type Ca2+ channels are regulated by TP. In vascular myocytes, the Ca2+ current is reduced by PTK phosphorylation of the Cav1.2b24,25 . PDGF stimulates tyrosine phosphorylation of multiple proteins and enhances L-type Ca2+ current21,22. Additionally, Cav1.2b co-immunoprecipitates with c-Src. PDGF regulation of current is inhibited by dialysis with antibodies to FAK or Src23. Cav1.2b is also regulated by at least three integrins. Whole cell recordings from single arteriolar myocytes show that soluble (nonclustering) ligands of αvβ3-integrin, such as RGD peptides, inhibit L-type Ca2+ current24. In contrast, soluble ligands of the laminin receptor, α4β1, enhance current25. Definitive evidence for regulation of Cav1.2c by the growth factor IGF-1, through Src, was recently presented. In cerebellar granule neurons, Cav1.2 unit is tyrosine phosphorylated in response to IGF-1, resulting in potentiation of L-type Ca2+ current26.
K+ Channels
K+ channels are the targets of many intracellular signaling pathways that include protein phosphorylation and dephosphorylation. The expression of cloned ion channels in heterologous host cells provides a convenient means of analyzing the molecular details of channel modulation27, 28. In the case of voltage-dependent K+ (Kv) channels, a consistent pattern has begun to emerge. The activity of several different cloned Kv channels is suppressed after phosphorylation by both receptor and nonreceptor tyrosine kinases29,30. The Kv channels are composed of tetramers of transmembrane α-subunits associated with up to four β-subunits. There are several Kv subfamilies that were reported to be TP controlled7,31.
The Kv1 family is affected directly by TP. In particular the activity of Kv1.2 was suppressed by the TK PYK2 while EGFR and Src suppressed the Kv1.5 channel, as well as by activation of several growth factor receptors30,32.
It has been shown that treatment with either pervanadate or insulin suppresses Kv1.3 current. Mutational analysis demonstrates that at least two distinct tyrosine residues are required for current suppression by pervanadate. Insulin receptor suppressed the current by TP of Kv1.3 by different combinations of tyrosine residues. In addition, TP of the Kv1.3 channel by EGFR produces an acceleration of C-type inactivation kinetics during patch clamp recording, while insulin does not alter this kind of inactivation. Kv1.3 is also phosphorylated directly by several nonreceptor TK that suppress the channel activity and modulate the channel kinetics. These findings point to a complex pattern of phosphorylation that modulates the suppression of Kv1.3 and involves receptors and non receptor TK3,33,34.
Inwardly rectifying K+ (Kir) channels are also regulated by TP. Perorthovandate a PTPs inhibitor, inhibited the current of Kir2.1 and Genistein reversed this suppression. These effects are prevented by site mutation of a tyrosine consensus residue for TK phosphorylation in the C-terminal domain of Kir2.1. After co-expression of nerve growth factor receptor with Kir2.1 channels in tsA-201 cells and Xenopus oocytes, the activity of Kir2.1 was rapidly suppressed by NGF. Acute inhibition was also evoked by EGF and insulin via endogenous insulin receptors, indicating that Kir2.1 channels may serve as a general target for neurotrophic growth factors in the brain35,36. Other members of Kir2 contains a Tyr residue at an equivalent position, so receptor PTKs have the potential to regulate other Kir2 channels by this mechanism37.
Na+ Channels
Both voltage-gated Na+ (VGNa) and Epithelial Na+ (ENaC) ion channels are subject to modulation by phosphorylation, and different channel properties are modulated depending on the specific type of channel and kinase involved. Modification of Na+ channel by TP was demonstrated in the PC12 neuronal cell line. Growth factors such as NGF, PDGF, EGF and FGF similarly and significantly inhibited VGNa current. The PTK inhibitors Tyrphostin AG9 and Tyrphostin AG876 reversed this acute inhibition. Moreover, suppression of current by the growth factors was not additive, indicating that a common signaling pathway is involved38,39. On the other hand, in ENaC Serine/threonine phosphorylation by PKA is a decisive trigger that converts the channels from voltage independent into partially voltage activated state. A large body of studies illuminates the role of growth factors on EnaC expression in epithelial cells, but evidence for direct regulation of ENaC by TP is not established yet40,41.
In Conclusion
It is very clear that tyrosine phosphorlyation converges on ion channels to fine-tune their activity. Although the physiological relevance of TP regulated ion channels is not well understood as yet, further investigation will provide an insight into the role that ion channels play as part of the whole cell signaling mosaic.
References
- Niklinska, B.B. et al. (1992) J. Biol. Chem. 267, 7154.
- Gould, E.M. et al. (1995) Am. J. Physiol. Cell. Physiol. 268, C1425.
- Toma, C. et al. (1995) Br. J. Pharmacol. 114, 1266.
- Wijetunge, S. et al. (2000) Br. J. Pharmacol. 129, 1347.
- Cataldi, M. et al. (1996) J. Biol. Chem. 271, 9441.
- Hsu, Y.T. and Molday, R.S. (1993) Nature 361, 76.
- Huang, X.Y. et al. (1993) Cell 75, 1145.
- Nakatani, K. et al. (1995) J. Physiol. 484, 69.
- Kaupp, U.B. and Seifert, R. (2002) Physiol. Rev. 82, 769.
- Gordon, S.E. et al. (1992) Neuron 9, 739.
- Molokanova, E. et al. (1997) J. Neurosci. 17, 9068.
- Molokanova, E. et al. (1999) J. Neurosci. 19, 4786.
- Savchenko, A. et al. (2001) PNAS U. S. A. 98, 5880.
- Molokanova, E. et al. (2003) J. Physiol. 552.2, 345.
- Krajewski, J.L. et al. (2003) J. Neuroscience 23, 10100.
- Marchetti, C. and Brown, A.M. (1988) Am. J. Physiol. 254, C206.
- MacEwan, D.J. and Mitchell, R. (1991) FEBS Lett. 291, 79.
- Nastainczyk, W. et al. (1987) Eur. J. Biochem. 169, 137.
- Hell, J.W. et al. (1993) J. Biol. Chem. 268, 19451.
- Slomiany, B.L. et al. (1992) Gen. Pharmacol. 23, 799.
- Gaetano Lombardi, G. et al. (1996) J. Biol. Chem. 271, 9441.
- Strauss O. et al. (1997) FASEB J. 11, 859.
- Liu, H. and Sperelakis, N. (1997) Can. J. Physiol. Pharmacol. 75, 1063.
- Hu, X.Q. et al. (1998) J. Biol. Chem. 273, 5337.
- Wijetunge, S. et al. (1992) Biochem. Biophys. Res. Commun. 189, 1620.
- Wu, X. et al. (1998) J. Cell Biol. 143, 241.
- Waitkus, K.R. et al. (1998) FASEB J. 12, A384.
- Blair, LA, and Marshall J. (1997) Neuron 19, 421.
- Kues, W.A. and Wunder, F. (1992) Eur. J. Neurosci. 4, 1296.
- Prevarskaya, N.B. et al. (1995) J. Biol. Chem. 270, 24292.
- Tsai, W. et al. (1997) EMBO J. 16, 4597.
- Dolly, J.O. et al. (1994) Biochem. Soc. Trans. 22, 473.
- Jan, L.Y. and Jan Y.N. (1997) Annu. Rev. Neurosci. 20, 91.
- Fadool, D.A. et al. (1997) J. Neurophysiol. 78, 1563.
- Bowlby, M.R. et al. (1997) J. Gen. Physiol. 110, 601.
- Fadool, D.A. and Levitan, I.B. (1998) J. Neurosci. 18, 6126.
- Wischmeyer, E. et al. (1998) J. Biol. Chem. 273, 34063.
- Pearson, R.B. and Kemp B.E. (1991) Methods Enzymol. 200, 62.
- Rogalski, S.L. et al. (2000) J. Biol. Chem. 275, 25082.
- Hilborn, M.D. et al. (1998) J. Neurosci. 18, 590.
- Tilly, B.C. et al. (1993) J. Biol. Chem. 268, 19919.