Voltage-dependent potassium channels (KV4) are expressed in the brain and the heart where they regulate cognitive function and cardiac output. Aging is associated with increased tendency for cardiovascular disease and cognitive decline but the link between those debilitating diseases is missing. Here we show that KV4 channels could possibly tie cardiovascular diseases and neuronal dysfunction and demonstrate a collection of functional tools from Alomone Labs to investigate this possibility.
Introduction
Structure
Potassium channels are a large and diverse protein superfamily that are typically grouped based on their structure and mode of activation23. The voltage-gated potassium (KV) channels encompass a large portion of this protein superfamily and are further divided into 12 subfamilies based on sequence homology to their Drosophila orthologs (Shaker, Shab, Shal and Shaw)18,23. The Shal-type potassium channels also known as KV4.1, KV4.2 and KV4.3 (in mammals) are highly expressed in the brain and the heart where they regulate numerous physiological functions including neuronal excitability and cardiac pacemaking respectively2. In the context of aging and its related diseases this article will mainly focus on these types of potassium channels.
The Shal-type potassium channels, share a prototypical structure with other potassium channels such as the cytoplasmic carboxy terminal, T1 assembly domain, a six α-helical transmembrane domain (S1-S6) and the pore loop (P-Loop), which is selective for potassium ions2. The helical S4 segment is unique among other S subunits as it is considered to be the voltage sensor due to dense clusters of positively charged arginine and lysine residues2. Upon membrane depolarization, the S4 domain mediates protein conformational change and opens the channel for potassium ion flux18.
The regulation over KV channel activity is a complex and multi-step process that depends on many factors including protein-protein interactions, cell signaling events, transcriptional and post translational activities as will be demonstrated in detail below.
Transcriptional regulation
KV4 expression is influenced by numerous factors including changes in transcriptional activity. Argenziano et al. found that testosterone controls KV4.3 expression in the heart1. Using Anti-KV4.3 Antibody (#APC-017), the authors showed that Finasteride and Flutamide (drugs preventing androgen signaling through different mechanisms) decrease KV4.3 expression in the right ventricle of rat heart, suggesting a role for testosterone and androgen signaling in potassium channel expression1.
Micro RNA (miRNA) miR-223-3p is highly expressed/upregulated in a rat model of acute myocardial ischemia (AMI)12. Its expression was shown to be inversely proportional to that of KV4.2 as determined by western blot analysis using Anti-KV4.2 Antibody (#APC-023). The possibility that miR-223-3p negatively regulates KV4.2 expression was tested and validated by expressing the miRNA in primary neonatal rat ventricular myocytes. Indeed, KV4.2 suppression by miR-223-3p was abolished by the administration of Antagomir to silence miR-223-3p12. The effect of miR-223-3p seemed to be specific to KV4.2 as protein levels of other KV channels, namely KV4.3, were not altered by the miRNA12.
In the nervous system cleaved GLP-1 peptide stimulates long term potentiation (LTP) signal in hippocampal nerves5. Western blot analysis of hippocampal lysates using Anti-KV4.2 Antibody showed decreased expression of the protein following chronic administration of GLP-1 (9-36) in mice which was associated with increased LTP signals5.
Subcellular Trafficking and Localization
The spatial distribution of potassium channels is important for proper cellular functions10. This is especially true for cells with complex structures such as neurons10. The possible role of TRPC6 channel in regulating KV4 channel distribution was recently addressed. TRPC6 knockdown resulted in a decrease of cell membrane associated KV4.3 and an increase in cytosolic abundance of the protein9 as evident by western blot of cytosolic/membrane fractions of hippocampal tissue homogenates using Anti-KV4.3 Antibody9. In addition, immunohistochemical staining of rat brain sections using Anti-KV4.3 Antibody showed that rats administered with TRPC6 siRNA displayed reduced KV4.3 clusters in dentate gyrus cells and parvalbumin (PV) positive GABAergic interneurons (Figure 1)9, supporting the possible role of TRPC6 in regulating KV4.3 subcellular distribution.
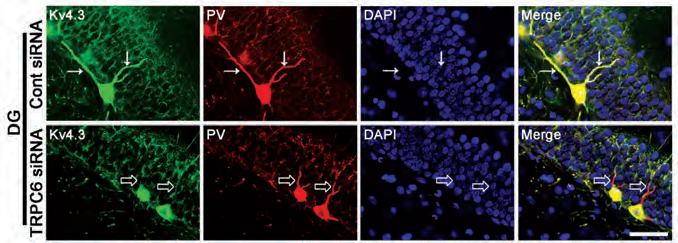
Immunohistochemical staining of rat brain sections using Anti-KV4.3 Antibody (#APC-017). KV4.3 staining (green) under control conditions (upper panels) is strongly expressed in GABAergic neurons and co-localizes with parvalbumin immunostaining (red). In response to TRPC6 siRNA administration (lower panels), KV4.3 distribution in dendrites significantly decreases.
Adapted from reference 9 with permission of Frontiers.
A novel interaction between the glycoprotein Nectin-2α and KV4.2 was shown to tune KV4.2 localization to specialized plasma membrane regions of adjacent somata of cholinergic neurons. This interaction was demonstrated by high resolution electron microscopy using Anti-KV4.2 Antibody16. Furthermore, genetic ablation of Nectin-2α in mice reduced KV4.2 fluorescent signal at the apical membrane of cholinergic neurons, confirming that Nectin-2α-KV4.2 interaction is important for KV4.2 localization.
KV4 Regulation by Post-Translational Modifications
Glycosylation is a common post translational modification important for numerous biological processes including protein folding in the endoplasmic reticulum (ER) and protein trafficking to the plasma membrane2. Recently, Endie et al., investigated the effects of syalic acid (a negatively charged sugar) modifications on KV channels in mouse ventricular myocytes6. Using transgenic mice lacking the sialyltransferase ST3Gal4, the authors demonstrated a delayed potassium outward (Ito) current that consequently delayed ventricular repolarization period as evident by prolonged QT intervals6. To investigate potential mechanisms, the authors used Anti-KV4.2 Antibody and Anti-KV1.5 (KCNA5) Antibody (#APC-004) to compare protein expression by western blot. Whereas, no change in protein expression was evident, the authors suggest that sialic acid modification of KV channels is a key step in finetuning KV channels activity6.
KV4 Interacting Proteins
The diversity in KV channel activity can be influenced by protein-protein interactions2. For example, Turnow et al., demonstrate that KV4.3- DPP10a interaction regulates transient potassium currents (Ito)21. Using immunofluorescence, they provide evidence for KV4.3-DPP10a co-localization using Anti-KV4.3 Antibody in human atrial myocytes and Chinese Hamster Ovaries (CHO)21. Furthermore, using functional assays they demonstrate this interaction is physiologically relevant since potassium currents were not visualized in the absence of DPP10a in CHO21.
Similarly, Wang et al., demonstrated the interaction between KV4.2-KChIP3- DPP10a in rat neocortical brain sections and olfactory bulbs using Anti- KV4.2 Antibody and suggest this protein complex mediates sub-threshold potassium currents in vivo22.
KV4 Channels in Cardiovascular Diseases
Potassium channels regulate the electrical driving force for normal cardiac function7. In particular, they act to restore membrane polarization and counterbalance depolarizing ions such as sodium and calcium7.
Impaired cardiac electrical activity is frequently observed following myocardial hypertrophy, which is an adaptive response to cardiac dysfunction. M3 muscarinic receptors are members of cholinergic receptors that innervate cardiac cells and control their electrical function. Chen et al., examined whether M3 over-expression could alleviate the adverse electrical signal after cardiac hypertrophy3. To test this hypothesis the authors generated transgenic mice, over-expressing M3 muscarinic receptors. Using Anti-M3 Muscarinic Receptor Antibody (#AMR-006), the authors confirmed the over-expression of M3 receptors by western blot and observed that that cardiac electrical activity was comparable to sham controls following transverse aortic constriction model3. To identify a potential mechanism by which M3 over-expression restores cardiac function, the authors examined the expression of various channels, which control potassium outflow and hence could potentially re-establish normal cardiac pace3. They found that M3 muscarinic receptors elevated Kir2.1 expression but no change was observed in KV4.3, as determined using Anti-KV4.3 Antibody3.
Recently, a genetic survey in a small cohort of Chinese population identified a missense mutation within KV4.3 gene (KCND3) associated with atrial fibrillation (AF)8. This mutation leads to Thr 361 to Ser (Thr→Ser) replacement in KV4.3 protein. To delve deeper into the mechanisms leading to atrial fibrillation, Huang et al., expressed wild type KV4.3 or its mutated counterpart, together with KChIp2 in HEK293T cells. Using this system, they discovered that Thr→Ser mutation increases total KV4.3 expression and increases its membrane localization, as determined by western blot with Anti-KV4.3 Antibody (Figure 2). In addition, the authors measured potassium currents and discovered that Thr→Ser is associated with KV4.3 gain of function8.
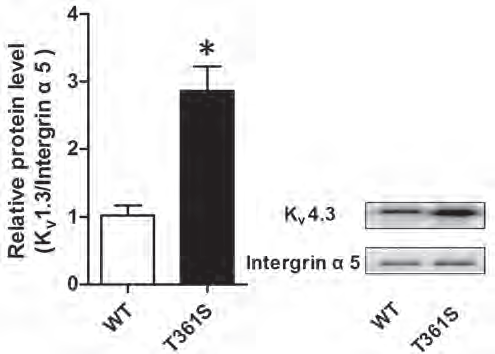
Western blot analysis of HEK 293 cells transfected with wildtype (WT) KV4.3 or with KV4.3 (T361S) mutant. Immunodetection of cell surface KV4.3 with Anti-KV4.3 Antibody (#APC-017) shows that the mutant displays increased cell-surface expression compared to WT. Integrin α5 is used as a loading control.
Adapted from reference 8 with permission of Impact Journals.
In contrast, Cheng et al., demonstrated that KV4.3 over-expression in cardiac myocytes protected mice from heart failure4. Specifically, using Anti-KV4.3 Antibody, the authors showed the association between elevated KV4.3 expression and decreased phosphorylation of calmodulin-dependent protein kinase (CaMKII) and suggested this protective effect to be due to calcium homeostasis4.
Recent studies have uncovered novel mutations within KV interacting proteins that can have debilitating effects on cardiac function such as the Brugada syndrome, which causes irregular heart beats7. Accordingly, Portero et al., identified a mutation in KVβ2 associated with the syndrome14. Using an in vitro expression system, the authors were able to confirm that Arg to Gln replacement does not affect KV4.3 expression as seen by western blot using Alomone Labs’ respective antibody, but rather affects cardiac electrophysiology in a manner that has yet to be uncovered.
Similarly, Tsai et al., identified KChIP1 copy number variants to be a strong genetic predictor for AF in Taiwanese population20. To investigate how KChIP1 is involved in AF, the authors undertook protein-protein interaction studies in adult rat hearts using Anti-KChIP1 (KCNIP1) Antibody (#APC-141) in pull-down assays and re-probing with antibodies against the potassium channels KV4.2 and KV4.3 or the calcium channel with Anti-CaV1.2 (CACNA1C) Antibody (#ACC-003)20. Unexpectedly, none of these proteins were detected, indicating a different mechanism by which KChIP1 regulates cardiac performance. The authors tried a genetic approach in which they silenced KChIP1 in atrial cell line, HL-1 and found a significant change in potassium currents and membrane depolarization, suggesting that KChIP1 itself regulates potassium outward currents20.
KV4 Channels in Neuronal Dysfunction
Cognitive decline due to aging is associated with CA1 and CA3 pyramidal neuron malfunction in the hippocampus17. Simkin et al., speculated that the increased firing in CA3 pyramidal neurons contributing to cognitive decline in aged brains may be caused by KV channels17. Administration of Phrixotoxin-1 (#STP-700), a selective and potent blocker of KV4.2 and KV4.3 channels significantly improved the electrophysiological activity of CA3 neurons in old rats (Figure 3). Additional results indicated that KV4.2 inhibition could possibly slow down neurodegenerative processes during aging17.
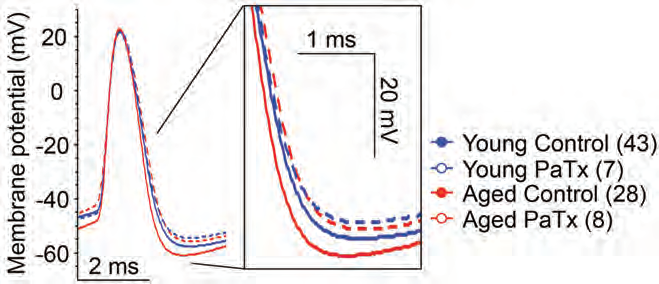
Representative traces of single orthodromically elicited action potentials from aged (red) and young (blue) CA3 neurons with (dashed line) and without (solid line) 1 µM Phrixotoxin-1 (#STP-700), (PaTx) in recording pipette (from AP threshold). PaTx treatment reduced the fast afterhyperpolarization (fAHP) of aged CA3 neurons to young-like values.
Adapted from reference 17 with permission of the Society for Neuroscience.
Parkinson’s Disease is another example of progressive neurodegenerative process where α-synuclein accumulation can harm vulnerable neurons such as the substantia nigra (SN) dopaminergic neurons19. In genetically engineered mice, bearing A53T mutation in α-synuclein protein, selective high firing rates in SN dopaminergic neurons (DA) were observed19. Comparable with the in vivo model, isolated neurons from mutated α-synuclein DA neurons displayed increased firing rates in a pattern that suggested a shift in pacemaker currents. Voltage-gated KV4 channels were previously shown to control dopaminergic neuron pacemaking. Application of Phrixotoxin-2 (#STP-710), a specific KV4 channel blocker completely hindered the difference in electrophysiological recordings between the control group and α-synuclein mutant neurons (Figure 4A)19. In addition, immunohistochemical staining of mouse brain sections using Anti-KV4.3 Antibody showed that KV4.3 expression is higher in substantia nigra of mice bearing the mutation in α-synuclein (Figure 4B). This suggests that altered KV4 activity or expression underlies this phenotypic difference19.
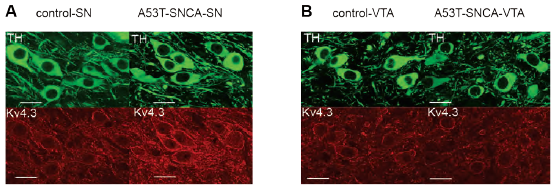
Immunohistochemical staining of mouse brain sections using Anti-KV4.3 Antibody (#APC-017). A. Expression of KV4.3 increases in DA substantia-nigra neurons of α-synuclein mutant mice (lower right panel). B. Expression of KV4.3 does not increase in DA neurons of the ventral tegmental area in α-synuclein mutant mice (lower right panel).
Adapted from reference 19 with permission of the Society for Neuroscience.
Different cell types respond differently to elevated α-synuclein levels11. For example, the dorsal motor nucleus of the vagus (DMV) nerve can tolerate high levels of α-synuclein levels and display milder apoptosis compared to the SN dopaminergic neurons11. Furthermore, α-synuclein accumulation alters electrical activity in SN dopaminergic neurons, but has no evident effect on DMV neurons11. In accordance, immunohistochemical staining of mouse brain sections bearing the A53T mutation in α-synuclein protein shows similar KV4.3 levels and expression pattern when compared to wildtype11.
Pharmacological Approaches to Studying KV4 Channels
Over the past decade, the list of small molecule/peptide inhibitors that target KV4 channels has grown substantially23. These tools, can now be exploited to discover novel functions and biological processes in which KV4 channels are involved. For example, Phrixotoxin-1 and AmmTx3 Toxin (#STA- 305) peptide toxin blockers for KV4.3 and KV4.2 were used to study how toxin transient potassium currents affect preBotzeiger type-1 neurons rhythmicity and their impact on breathing13.
In a similar way Heteropodatoxin-2 (#STH-340), a specific KV4.2 blocker was used to uncover a novel mechanism of synaptic plasticity in tuft dendrites of layer 5 pyramidal neurons15 where low frequency electrical stimulation in tuft dendrites induced long term potentiation activity.
References
- Argenziano, M. et al. (2017) J. Physiol. Sci. 67, 217.
- Birnbaum, S.G. et al. (2004) Physiol. Rev. 84, 803.
- Chen, X. et al. (2017) Cell. Physiol. Biochem. 43, 915.
- Cheng, J. et al. (2017) Oncotarget 8, 104037.
- Day, S.M. et al. (2017) Hippocampus 27, 1264.
- Ednie, A.R. and Bennett, E.S. (2015) J. Biol. Chem. 290, 2769.
- Giudicessi, J.R. and Ackerman, M.J. (2012) Nat. Rev. Cardiol. 9, 319.
- Huang, Y. et al. (2017) Oncotarget 8, 115503.
- Kim, J.E. et al. (2017) Front. Cell Neurosci. 11, 413.
- Lai, H.C. and Jan, L.Y. (2006) Nat. Rev. Neurosci. 7, 548.
- Lasser-Katz, E. et al. (2017) J. Neurosci. 37, 47.
- Liu, X. et al. (2016) Cell. Physiol. Biochem. 39, 102.
- Phillips, W.S. et al. (2018) J. Neurosci. 38, 3039.
- Portero, V. et al. (2016) J. Am. Heart Assoc. 5, e003122.
- Sandler, M. et al. (2016) Neuron 90, 1028.
- Shiotani, H. et al. (2018) J. Comp. Neurol. 526, 1527.
- Simkin, D. et al. (2015) J. Neurosci. 35, 13206.
- Snyders, D.J. (1999) Cardiovasc. Res. 42, 377.
- Subramaniam, M. et al. (2014) J. Neurosci. 34, 13586.
- Tsai, C.T. et al. (2016) Nat. Commun. 7, 10190.
- Turnow, K. et al. (2015) Basic Res. Cardiol. 110, 5.
- Wang, W.C. et al. (2015) J. Comp. Neurol. 523, 608.
- Wulff, H. et al. (2009) Nat. Rev. Drug Discov. 8, 982.