Disease outcomes diverge when astrocytes activate distinct G-protein coupled receptors (GPCR)-defined signaling pathways with opposing effects on neuronal excitability.
Astrocyte reactivity is a consistent feature of several neurological diseases, including epilepsy, Alzheimer’s disease, stroke, and neuropathic pain. But for years, disease-associated astrocytes were frequently grouped into a single reactive category when interpreting their effects on neural circuits. They were characterized as ‘reactive’, exhibiting abnormal calcium signaling, and are assumed to broadly amplify pathology. This description has been useful, but it is not complete.
Reactive astrocytes are not molecularly uniform. Across diseases, different astrocyte subtypes upregulate different receptors, activate different downstream pathways, and interact with neurons in distinct ways. What has remained unclear is whether these differences matter functionally. Do some astrocyte states actively push circuits toward pathology, while others work to pull them back?
Two recent studies answer that question directly. By examining astrocyte-specific G-protein coupled receptors (GPCR) pathways and testing their causal effects on neural circuits, it was revealed that astrocytes function as directional regulators of disease. Specifically, astrocytes are capable of either driving hyperexcitability or enforcing homeostasis, depending on which signaling pathway is engaged.
When Astrocytes Push Circuits Toward Hyperexcitability
In multiple neurological diseases, including epilepsy and stroke, astrocytes upregulate the purinergic Gq-coupled P2Y1 receptor (P2Y1R). But correlation alone cannot establish function. To test causality, Shigetomi and colleagues took injury out of the equation entirely (1).
In a middle cerebral artery occlusion (MCAO) stroke model in mice, they selectively overexpressed P2Y1R only in astrocytes [confirmed via immunohistochemistry with Alomone’s knockout-validated Anti-P2Y1 Receptor Antibody (#APR-009)], leaving neurons untouched and thus avoiding inflammation or tissue damage (Figure 1). Yet neuronal circuits became hyperexcitable anyway. Hippocampal electroencephalogram (EEG) recordings revealed increased epileptiform-like spikes and chemically induced seizures occurred at lower thresholds. At the single-cell level, neurons fired more action potentials in response to the same depolarizing input.
These effects were not explained by changes in basic membrane properties or baseline synaptic strength. Instead, astrocytes were actively reshaping circuit responsiveness.
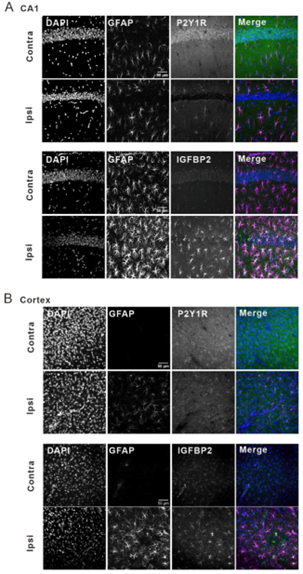
Figure 1. P2Y1R and IGFBP2 are co-upregulated in reactive astrocytes in the cortex and the hippocampus of MCAO model mice. Immunohistochemistry of P2Y1R and IGFBP2 in reactive astrocytes in (A) the hippocampal CA1 region (CA1) and (B) the cortex of MCAO model mice.
Image adapted from Shigetomi et al. (2024). https://doi.org/10.1038/s41467-024-50190-7. Licensed under Creative Commons Attribution 4.0 International (CC BY 4.0).
Astrocytic P2Y1R overexpression dramatically enhanced astrocyte calcium signaling. Using dual-color two-photon imaging, the authors showed that astrocytic calcium responses became larger, spread across entire astrocyte territories, and were tightly coupled to neuronal activity.
But calcium itself was not the endpoint. The key question became: what do astrocytes do with this signal?
Transcriptomic analysis provided the answer. Astrocytic P2Y1R activation upregulated insulin-like growth factor binding protein 2 (IGFBP2). IGFBP2 acted as an excitatory signal, enhancing neuronal activity through IGF-1 receptor signaling. Importantly, reactive astrocytes in epilepsy and stroke models showed coordinated upregulation of both P2Y1R and IGFBP2.
This established a defined push pathway: astrocytic P2Y1R activation → IGFBP2 release → increased neuronal excitability. Thus, astrocytes were actively driving circuit instability.
When Astrocytes Pull Circuits Back Toward Homeostasis
A very different astrocyte story emerges from the spinal cord. Xu and colleagues began not with pathology, but with identity. Using single-nucleus RNA sequencing, they identified GPR37L1 as one of the most enriched GPCRs in spinal dorsal horn astrocytes (2). Unlike P2Y1R, which rises in disease, GPR37L1 marked a specific astrocyte population present under baseline conditions.
Protein-level validation was essential here. The authors confirmed astrocytic localization of GPR37L1 using Alomone’s Anti-GPR37L1 (extracellular) Antibody (#AGR-050), and the specificity of GPR37L1 immunostaining was validated in GPR37L1 knockout (KO) mice.
To illustrate receptor localization at the protein level, representative immunostaining using Anti-GPR37L1 Antibody from Alomone Labs (#AGR-050) is shown in Figure 2.
What happens when this receptor is lost?
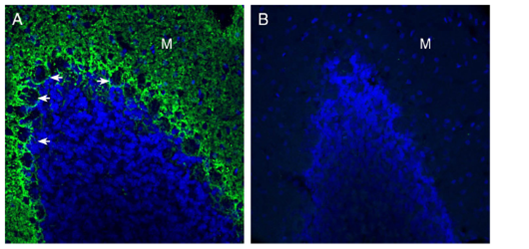
Figure 2. Expression of GPR37L1 in the rat cerebellum. Immunohistochemical staining of perfusion-fixed frozen rat brain sections with Anti-GPR37L1 (extracellular) Antibody (#AGR-050), (1:300), followed by goat anti-rabbit-AlexaFluor-488. (A) GPR37L1 immunoreactivity (green) appeared around Purkinje cell soma (arrows) and in the molecular layer (M). (B) Pre-incubation of the antibody with GPR37L1 (extracellular) Blocking Peptide (BLP-GR050), suppressed staining. Cell nuclei were stained with DAPI (blue). In-house staining by Alomone Labs.
Mice lacking GPR37L1 developed neuropathic pain faster after nerve injury, and failed to recover. Moreover, pain hypersensitivity persisted in GPR37L1-deficient mice long after wild-type animals returned to baseline. Even more telling, selectively knocking down GPR37L1 only in astrocytes, in otherwise healthy animals, was sufficient to induce pain hypersensitivity.
Electrophysiology revealed the circuit-level consequences of the absence of GPR37L1. Excitatory synaptic transmission in the spinal dorsal horn was amplified. Spontaneous excitatory postsynaptic currents increased in both frequency and amplitude. Astrocytes became reactive, but microglia did not—placing astrocytes squarely at the center of the effects.
The pathology was not attributed to astrocytic activation, instead there was a loss of an astrocyte-intrinsic restraining mechanism. Mechanistically, GPR37L1 regulated excitatory transmission through the astrocytic glutamate transporter-1 (GLT-1). Loss of GPR37L1 reduced GLT-1 expression, impairing glutamate clearance. Overexpression of GPR37L1 restored GLT-1 levels and normalized synaptic activity. The specificity of this effect mattered. Expression of another astrocytic transporter, GABA Transporter 3 (GAT-3), remained unchanged when GPR37L1 was knocked down, suggesting that GPR37L1 regulates excitatory synaptic transmission and neuropathic pain through its association with GLT-1. GAT-3 levels were confirmed using Alomone’s Anti-GABA Transporter 3 (GAT-3) Antibody (#AGT-003).
Super-resolution imaging and proximity ligation assays showed that GPR37L1 and GLT-1 were found in close spatial association within astrocytic processes. Activation of GPR37L1 by maresin-1 increased astrocytic glutamate uptake, directly linking receptor signaling to transporter function.
This defined a pull pathway: astrocytic GPR37L1 signaling → GLT-1–mediated glutamate clearance → reduced excitatory drive. When this pathway was intact, circuits stabilized. When it was lost, hyperexcitability persisted.
A Push-pull Model of Astrocyte Control
Taken together, these studies reposition astrocytes from a single reactive state to a push–pull system that governs circuit direction: P2Y1R/IGFBP2 signaling pushes circuits toward hyperexcitability and seizure susceptibility, while GPR37L1/GLT-1 signaling pulls circuits back toward homeostasis and enables recovery from neuropathic pain.
In both cases, astrocytes are active participants. What differs is not whether they are reactive, but which molecular pathway they engage. These papers elegantly show that astrocytes are directional, and in disease, direction matters.



 Antibody")
 Antibody")