Platelets and sensory neurons emerge as decision-makers in neuro-immune control.
For years, neuro-immune communication was discussed as a background condition: inflammation altered brain function, neural activity shaped immune tone, and the details were often treated as secondary. Two recent mouse studies push that view into the foreground. Together, they show that peripheral cells make active, quantitative decisions about how the brain adapts and how the immune system mounts antibody responses, using defined molecular levers rather than diffuse signaling. One study follows the path inward, the other traces it outward. What connects them is less about direction, and more about control.
Platelets as Regulators of Inhibitory Plasticity
In the first study, Garofalo and colleagues focused on fear memory – one of the most tightly regulated forms of learning in the mammalian brain (1). The hippocampus sits at the center of this process, and serotonin is a known modulator of hippocampal inhibition and plasticity. What had remained unclear was how peripheral physiology contributes to setting serotonin levels in a healthy brain. The answer, unexpectedly, turned out to involve platelets.
When the authors reduced platelet number or blocked platelet activation, hippocampal serotonin levels fell. Fear learning and contextual fear memory increased. In addition, long-term potentiation in the hippocampus strengthened. These changes occurred without altered activity in raphe serotonergic neurons, ruling out a central serotonin production effect. Restoring serotonin levels with administration of its precursor reversed both the synaptic and behavioral changes, directly linking serotonin availability to the fear and learning phenotype.
The missing step was how platelet serotonin handling is regulated upstream. That control is attributed to natural killer (NK) cells acting in the gut. NK cells set IL-13 levels, which influence serotonin production by enterochromaffin cells and serotonin uptake by platelets. To quantify this step, the authors measured surface expression of the serotonin transporter on platelets via flow cytometry using Anti-Serotonin Transporter (SERT) (extracellular)-FITC Antibody (#AMT-004-F). They found that NK cell depletion reduced platelet serotonin uptake capacity in parallel with the behavioral effects (Figure 1). The immune system, through a defined peripheral circuit, was tuning hippocampal inhibition and memory formation.
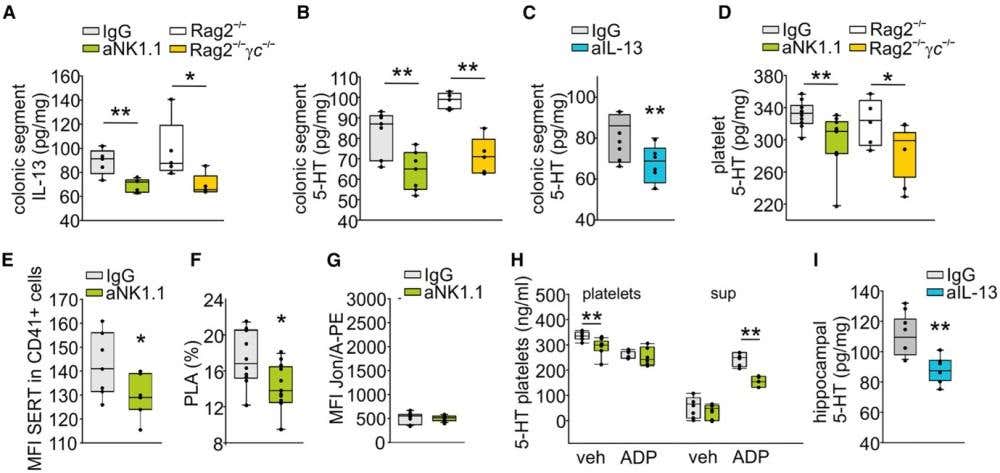
Figure 1. Natural killer (NK) cell-platelet dialogue. (A) ELISA analysis of IL-13 levels in the colonic segment of IgG- or aNK1.1-treated male mice or Rag2−/− and Rag2−/−γc−/−. (B) ELISA analysis of 5-HT levels in the colonic segment of IgG- or aNK1.1-treated male mice or Rag2−/− and Rag2−/−γc−/− mice. (C) ELISA analysis of 5-HT levels in the colonic segment of IgG- or IL-13-treated mice. (D) ELISA analysis of 5-HT content in the platelets sorted from IgG- or aNK1.1-treated male or Rag2−/− and Rag2−/−γc−/− mice. (E) Expression of SERT on the surface of platelets (CD41+ cells), expressed as the median fluorescence intensity (MFI) in mice treated with the NK-depleting antibody or the IgG isotype control antibody (n = 7, Student’s t-test, ∗p = 0.021). (F) Quantification of circulating platelet-leukocyte aggregates (PLAs) in whole blood of mice depleted or not depleted of NK cells, measured as the percentage of CD45+ leukocytes that were positive for the platelet-specific marker CD41 (unpaired two-tailed t test, ∗p = 0.05). (G) Activation of the platelet-specific integrin αIIbβ3 (JON/A) in untreated whole blood of mice depleted or not depleted of NK cells, expressed as the MFI of JON/A-PE, an antibody specific for the active conformation of integrin αIIbβ3. (H) ELISA analysis of 5-HT content in platelets and platelet medium (sup) sorted from IgG- or aNK1.1-treated mice and stimulated in vitro with vehicle (veh) or ADP (25 μM). (I) ELISA analysis of hippocampal 5-HT levels in IgG- or IL-13-treated. For boxplots, the center line, boxes, and whiskers represent the median, inner quartiles, and the remaining data distribution, respectively.
Image adapted from Garofalo et al. (2025) https://doi.org/10.1016/j.celrep.2025.115261. Licensed under Creative Commons Attribution 4.0 International (CC BY 4.0).
When Sensory Neurons Decide the Antibody Response
The second study moves in the opposite direction, asking how signals originating in the nervous system shape immune outcomes. Aguilar and colleagues addressed a long-standing gap in neuro–immune research: we know sensory neurons sense pathogens and regulate innate immunity, but their role in humoral immune responses in vivo was unclear (2).
Using mouse models of Streptococcus pneumoniae infection and Alternaria alternata-induced asthma, the authors showed that sensory neurons are required for effective B cell recruitment and antibody production. During pneumococcal recall infection, depleting TRPV1-expressing sensory neurons using resiniferatoxin (RTX), increased bacterial burden and reduced B cell numbers, IgG release, and neutrophil activation. In allergic airway inflammation, the same neuronal depletion reduced B cell populations, IgE levels, and asthmatic features. Sensory neurons were therefore necessary to mount antibody responses across distinct immune challenges.
The mechanism depended on stimulus-specific neuropeptide release. During bacterial infection, sensory neurons preferentially released vasoactive intestinal polypeptide (VIP). Loss of sensory neurons reduced VIP signaling, weakened humoral responses, and impaired bacterial clearance. Administering VIP to sensory neuron-depleted mice restored bacterial clearance, while genetic loss of VIP receptor 1 (VIPR1) increased infection severity. Flow cytometry using our Anti-VPAC1 (VIPR1) (extracellular)–FITC Antibody (#AVR-001-F) confirmed the engagement of VIP-responsive immune cell populations, supporting a direct role for VIP signaling in shaping humoral immunity (Figure 2).
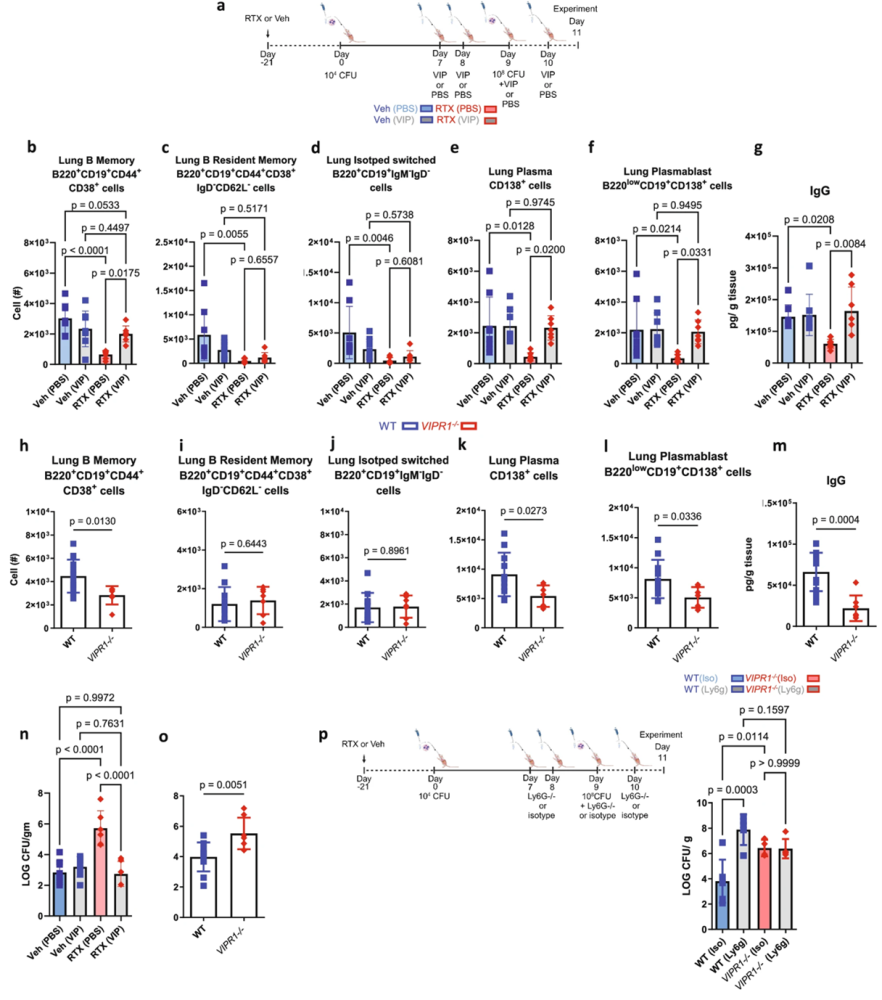
Figure 2. Vasoactive intestinal peptide (VIP) supplementation enhanced bacterial clearance, and increased the production of memory B cells, plasma cells, and IgG in response to pre-exposure and infection with Streptococcus pneumoniae. (a) A schematic of VIP or vehicle (Veh; PBS) delivery. The images of the model of S. pneumoniae pre-exposure and infection with serotype 19 F were created in BioRender. Aguilar, D. (2022) BioRender.com/h64o602. The cells were gated on live CD45+ cells, and total populations were assessed by counting beads. (b) B memory cells, (c) B resident memory cells, (d) isotype switched cells, (e) plasma cells, and (f) plasmablasts were reduced with sensory neuron ablation. Supplementation with VIP increased B cell populations. VIP supplementation did not further increase B cell populations in mice with intact sensory neurons. (g) IgG was reduced with RTX as previously described, but increased with the supplementation of VIP to RTX mice. (h) VIP1 receptor knockout (VIPR1–/–) mice had reduced B memory cells compared to wild-type (WT) mice. (i) B resident memory and, (j) Isotype switched B cells were similar in VIPR1–/– and WT mice. (k) Plasma cells and (l) plasmablasts were reduced in VIPR1–/– vs. WT mice. The data were analyzed with a two-sided T-test. WT n = 5, VIPR1–/– n = 5. (m) VIPR1–/– mice had reduced IgG following pre-exposure and infection compared to WT littermates. (n) VIP supplementation suppressed bacterial burden in RTX mice. No effect of VIP supplementation was observed in Veh mice. Veh (PBS): n = 8, Veh (VIP): n = 7-8, RTX (PBS): n = 6-7, RTX (VIP): n = 6-7. (o) VIPR1–/– mice have increased bacterial burden following pre-exposure and infection with S. pneumoniae compared to WT littermates. (p) Ly6g neutralizing antibody or isotype (Iso) antibody was given to WT or VIPR1–/– mice. The image was created in BioRender. Aguilar, D. (2022) BioRender.com/h64o602. The bacterial burden (Log CFU/g tissue mass) from lung homogenates was greater upon neutrophil depletion in WT mice compared to isotype control mice. VIPR1–/– mice did not further increase bacterial burden upon neutrophil depletion, demonstrating that VIP significantly stimulates B cells to increase neutrophil-mediated bacterial clearance.
Image adapted from Aguilar et al. (2024). https://doi.org/10.1038/s41467-024-53269-3. Licensed under Creative Commons Attribution 4.0 International (CC BY 4.0).
The same neurons initiated a different antibody program under allergic conditions. In airway inflammation, sensory neurons released substance P rather than VIP, promoting IgE production and disease pathology. Removing sensory neurons reduced IgE levels and inflammation, while reintroducing substance P reinstated these features. The neurons were not broadly amplifying immunity; they were selecting which antibody response to deploy based on the immunological context.
As in the platelet study, the conclusions rested on cellular specificity. Selective ablation of TRPV1-expressing sensory neurons was confirmed using our knockout-validated Anti-TRPV1 (VR1) Antibody (#ACC-030) in vagal ganglia. This was conducted to ensure that any changes in antibody responses reflected targeted disruption of neuropeptide-releasing sensory neurons rather than non-specific neural damage.
Together, the data show that sensory neurons actively regulate humoral immunity by releasing defined neuropeptides – VIP or substance P – that act on B cells in a stimulus-dependent manner.
A Distributed System for Learning and Immunity
Taken together, these papers describe the same architecture from different sides. Platelets adjust brain plasticity by setting serotonin availability, while sensory neurons instruct B cells by selecting context-appropriate neuropeptide signals. In both cases, the effects are precise, measurable, and reversible.
The shared implication is that neuro-immune communication is a distributed control system, implemented by specific peripheral cells that regulate signal handling at the level of transporters and peptides. Learning and immunity emerge from these decisions.
As interest in brain-body communication grows, these studies set a higher bar. They show how we need to understand more than just directionality; we also need to identify the cells that decide, the molecules they adjust, and the physiological consequences that follow.



 (extracellular)-FITC Antibody")
 (extracellular)-FITC Antibody")
 Antibody")