A look at pre- and postsynaptic molecular markers in neurodevelopmental and psychiatric disorders.
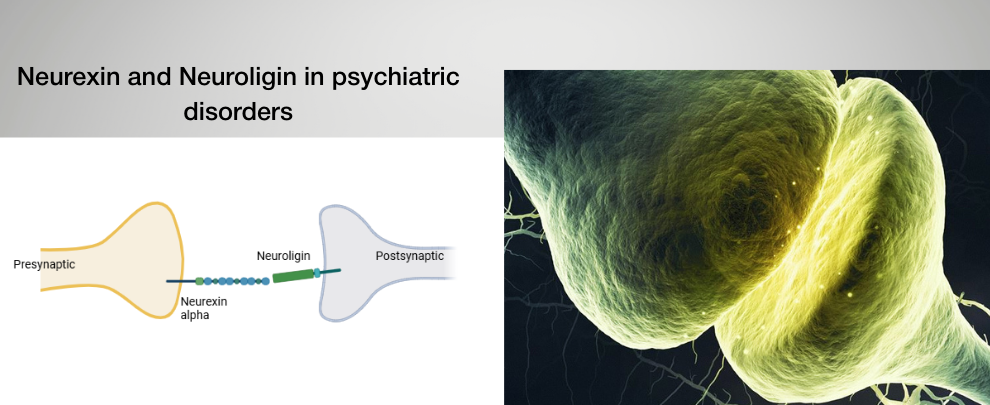
Neurexins and neuroligins are trans-synaptic adhesion molecules that form a polarity-defined molecular bridge across the synaptic cleft. These single-pass membrane proteins are positioned on opposite sides of the synapse: neurexins reside in the presynaptic membrane, where they organize vesicle release machinery, while neuroligins are embedded in the postsynaptic membrane, where they anchor scaffold proteins and neurotransmitter receptor complexes. Their extracellular domains bind across the cleft to align pre- and postsynaptic specializations, while their intracellular domains recruit distinct signaling and structural complexes. Because of this structural asymmetry, disruption on the presynaptic side is expected to affect release and adhesion, whereas disruption on the postsynaptic side should impair receptor anchoring and synaptic stabilization. Converging genetic and mechanistic evidence links this adhesion axis to neurodevelopmental and psychiatric disorders, suggesting that synaptic polarity—and which side of the bridge fails—may determine circuit-level and behavioral outcomes.
Here, we focus on the neurexin-neuroligin adhesion axis. Two recent studies independently perturbed opposite poles of the same trans-synaptic complex. Together, the research showed that disrupting pre- or postsynaptic adhesion nodes produced distinct but measurable circuit-level and behavioral phenotypes.
A Synapse Built on Polarity
The neurexin-neuroligin complex is structurally asymmetric: neurexin 1α (NRXN1α) resides presynaptically, organizing release machinery and binding postsynaptic neuroligins, while neuroligin 2 (NLGN2) resides postsynaptically at inhibitory synapses, anchoring gephyrin and γ-aminobutyric acid (GABA) receptor complexes.
This arrangement creates a polarity-defined bridge, where extracellular adhesion aligns pre- and postsynaptic membranes, while intracellular domains recruit scaffolds that stabilize transmission. Therefore, disturbing extracellular binding should destabilize contact across the cleft, whereas disrupting intracellular domains should weaken postsynaptic anchoring despite intact adhesion.
Two groups tested exactly these scenarios.
Postsynaptic Failure: an Neurologin 2 (NLGN2) C-Terminal Truncation Weakens Inhibition
A recent study focused on the following question: is the intracellular C-terminal domain of NLG2 required for inhibitory circuit stability (1)? To test this, they inserted a premature stop codon after residue L713 in the Nlgn2 gene, which deleted the final 123 amino acids of NLGN2. This mutation generated a “StopKI” mouse model. Since the NLGN2 C-tail mediates intracellular protein-protein interactions and posttranslational regulation, truncation should impair inhibitory synapse stabilization. Overall, this left the extracellular adhesion domain intact, thus NRXN binding should persist, but the intracellular signaling capacity would be compromised.
Electrophysiology confirmed the outcome. In hippocampal CA1 pyramidal neurons, miniature inhibitory postsynaptic currents were reduced. Moreover, immunohistochemistry showed decreased vGAT and gephyrin puncta density in CA1, as well as reduced GABAergic synaptic density. This allowed the researchers to deduce that inhibitory synaptic stabilization requires the NLGN2 C-tail. Behavior followed physiology. Both male and female StopKI mice exhibited increased obsessive-compulsive disorder (OCD)-like phenotypes as well as deficits in social cognition not previously reported in full NLGN2 knockout mice.
Rather than a global neuroligin loss, the researchers observed domain-specific signaling disruption. Here, Anti-Neuroligin 2 (extracellular) Antibody (#ANR-036) functioned as a quantitative postsynaptic marker. It was used for surface labeling (Figure 1), synaptic puncta analysis, and co-immunoprecipitation, helping to understand where postsynaptic structure failed.
The results demonstrated that if intracellular scaffolding collapses, intact adhesion is insufficient for maintaining surface expression of NLGN2, which results in a reduction of GABAergic synaptic density.
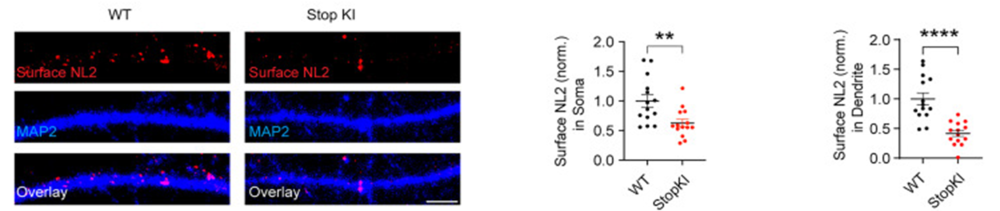
Figure 1. Surface Neuroligin 2 (NL2) staining was significantly reduced in cultured StopKI hippocampal neurons (DIV15–18). The left panel shows surface NL2 staining in wild-type (WT) and StopKI hippocampal neurons. The two right panels show the quantitation of surface NL2 in soma and dendrites, respectively (n = 14 cells for both WT and StopKI, from 3 independent cultures; Student’s t-test, NL2 in soma, t = 3.046; df = 21.90; **p = 0.0059; NL2 in dendrites, t = 5.149; df = 19.56; ****p < 0.0001). Scale bar, 5 µm. The error bars indicate the SEM.
Image adapted from Pandey et al. (2025). https://doi.org/10.1523/JNEUROSCI.1417-24.2025. Licensed under Creative Commons Attribution 4.0 International (CC BY 4.0).
Presynaptic Failure: Autoimmune NRXN1α Disrupts Adhesion and Excitation
The second study focused on NRXN1α from a different angle. Genetic studies implicated NRXN1α in schizophrenia risk, so the authors asked whether an autoimmune mechanism might target this presynaptic molecule (2).
Screening 387 schizophrenic patients identified anti-NRXN1α autoantibodies in 2.1% of cases and none were detected in 362 controls. Therefore, a definable antibody-positive subgroup of schizophrenic patients exists.
Biochemical assays showed that the patient autoantibodies inhibited NRXN1α binding to neuroligin 1 (NLG1) and NLG2. The disruption occurred extracellularly, at the adhesion interface, and functional consequences followed. In the mouse anterior cingulate cortex, the miniature excitatory postsynaptic currents (mEPSCs) frequency declined after exposure to patient IgG. Spine density also decreased, and mice developed impaired prepulse inhibition, reduced cognitive performance, and diminished social novelty preference. Removing anti-NRXN1α autoantibodies from the IgG fraction mitigated these deficits. Thus, it was clear that the autoantibodies were pathogenic.
Alomone’s Anti-Neurexin 1α (extracellular) Antibody (#ANR-031) was used throughout for Western blotting, immunohistochemistry, and immunoprecipitation (Figure 2). In these contexts, the antibody played two roles: structural markers of presynaptic adhesion and disease-causing effectors.
Here, the principle differs from the NLG2 study described earlier. Extracellular adhesion failure destabilizes excitatory transmission even when postsynaptic machinery remains intact.
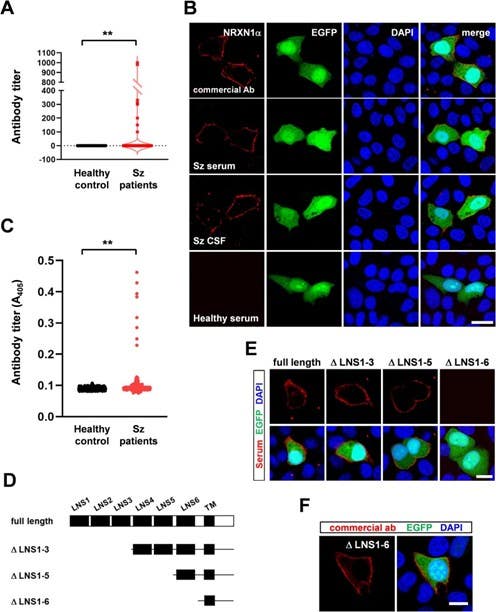
Figure 2. Identification of anti-neurexin 1α (NRXN1α) autoantibodies in schizophrenia (Sz) patients. A. Titers of serum levels of anti-NRXN1α autoantibodies quantitated with a cell-based assay. **p < 0.01 (N = 362 healthy controls; N = 387 patients with Sz; Mann–Whitney U-test). B. Immunocytochemistry using Alomone’s anti-NRXN1α antibody as control. The serum and CSF were used from Sz patient 1, and serum was used from healthy controls. C. Titers of serum levels of anti-NRXN1α autoantibodies quantitated by an enzyme-linked immunosorbent assay (ELISA). **p < 0.01 (N = 362 healthy controls; N = 387 patients with Sz; Mann–Whitney U-test). D. NRXN1α deletion constructs. E. Immunocytochemistry using serum from patient 1 with Sz, who was positive for anti-NRXN1α autoantibodies. NRXN1α deletion constructs and EGFP were expressed from a plasmid. Similar results were obtained from all anti-NRXN1α antibody-positive patients with Sz. Bar: 10 μm. F. Immunocytochemical confirmation of the expression of NRXN1αΔLNS1-6 using Alomone’s anti-NRXN1α antibody. Bar: 10 μm.
Image taken from Shiwaku, et al. (2023). https://doi.org/10.1016/j.bbi.2023.03.028. Licensed under Creative Commons Attribution 4.0 International (CC BY 4.0).
Adhesion Axis, Divergent Circuits
These two perturbations act on opposite poles of the same molecular bridge:
- Postsynaptic NLGN2 C-tail deletion → reduced inhibition → OCD-like phenotypes
- Presynaptic NRXN1α autoimmunity → reduced excitation → schizophrenia-like phenotypes
Both studies demonstrated how adhesion disruption altered synaptic transmission, which produced behavioral changes. But polarity matters – which side fails determines which circuit shifts.
The neurexin-neuroligin complex illustrates how we can use antibody markers to help measure synaptic physiology and link circuit imbalances or disruption to behavior. These results highlighted a clear principle, that pre- and postsynaptic nodes are mechanistically distinct, and experimentally separable, but behaviorally consequential.


 Antibody")
 Antibody")