Ion channels play central roles in cancer by regulating processes essential for tumor initiation and progression. By controlling the movement of Ca²⁺, K⁺, Na⁺, and Cl⁻ across cellular membranes, they shape membrane potential, intracellular signaling, cell cycle progression, and cell volume dynamics. In tumors, ion channel expression and activity are frequently dysregulated, contributing to sustained proliferation, resistance to apoptosis, metabolic adaptation, angiogenesis, and enhanced migratory and invasive behavior.
Beyond biochemical signaling, cancer progression is deeply influenced by mechanics. Cancer cells and their microenvironment are mechanically abnormal – tumors are crowded, stiff, and generate excessive force. These physical changes have multiple downstream effects and can reshape calcium signaling, cytoskeletal organization, inflammatory programs, and metastatic potential. A central question, however, is how mechanical forces are converted into specific, cancer-relevant cellular behaviors.
Mechanosensitive ion channels provide a compelling answer. Among them, TRPV4, a calcium-permeable cation channel responsive to membrane stretch, osmotic stress, matrix stiffness, and mechanical force, has emerged as a recurrent signaling node in tumors. TRPV4 not only senses mechanical inputs but translates them into intracellular dynamics that regulate cell volume, cytoskeletal tension, transcriptional programs, and cell-cell or cell-matrix interactions.
According to three recent studies focused on breast, pancreatic, or bladder cancer, TRPV4 increasingly appears as a central mechanotransduction hub that converts physical cues into biological outcomes. Notably, its functional consequences are context-dependent: in some settings, TRPV4 activity promotes migration, invasion, and pro-tumorigenic remodeling, while in others it constrains growth or alters differentiation programs in ways that may be anti-tumorigenic. These divergent effects likely reflect differences in tissue type, cellular identity, and the specific mechanical regimen encountered. Together, these findings positioned TRPV4 — and mechanosensitive ion channels more broadly — as key integrators of mechanics and signaling in cancer biology.
Mechanical Context Determines Whether TRPV4 Restrains or Enables Invasion
In high-grade breast ductal carcinoma in situ (DCIS), mechanical crowding promotes invasion only when TRPV4 activity is suppressed (1). Under overcrowded conditions, DCIS cells showed reduced intracellular Ca²⁺ transients, relocalization of TRPV4 away from the plasma membrane (Figure 1), loss of mechanically induced Ca²⁺ fluctuations, and a measurable reduction in cell volume. Alomone’s Anti-TRPV4 (extracellular) Antibody (#ACC-124) was essential for mapping the distribution of TRPV4 at the cell membrane. Pharmacologic inhibition or genetic silencing of TRPV4 reproduced this phenotype – cell shrinkage, diminished mechanosensitive Ca²⁺ signaling, and increased invasive escape – while TRPV4 activation increased cell volume, restored Ca²⁺ signaling, and suppressed invasion even under crowded conditions.
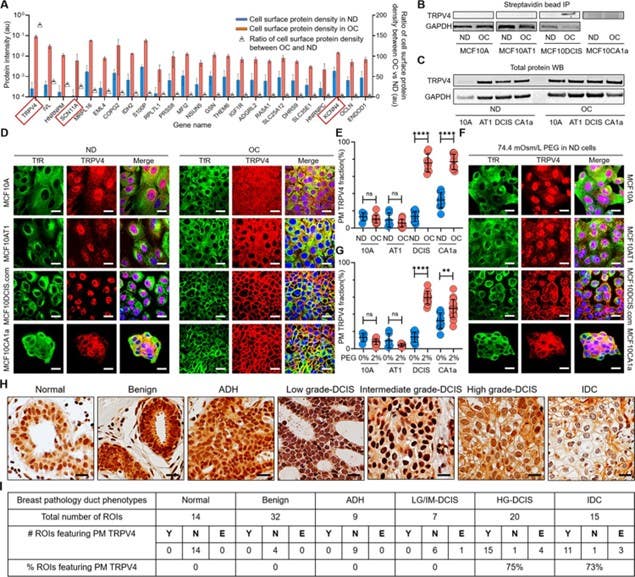
Figure 1. Cell crowding induces TRPV4 relocalization to the plasma membrane (PM) in MCF10DCIS.com cells. (A) Mass spectrometry data showing proteins enriched in the PM > 5-fold (fold changes represented using triangle plots) when cells are under overconfluence (OC) conditions (red bars) relative to normal density (ND) conditions (blue bars). The OC/ND ratio is on the right axis. Ion channels are marked with red boxes, where TRPV4 shows an approximate 160-fold increased association with the PM under OC conditions. Refer to (1) for the gene names. (B) Proteins near and on the PM were immunoprecipitated (IP) with streptavidin beads after cell surface biotinylation and were analyzed for TRPV4 by Western blotting (WB). TRPV4 was significantly associated with the PM in OC MCF10DCIS.com cells. In MCF10CA1a cells, TRPV4 appeared to be associated with the PM under both ND and OC conditions, with a slight increase under OC conditions. (C) WB of whole protein lysates demonstrated similar overall TRPV4 protein levels across MCF10A cell derivatives (10A, AT1, DCIS, and CA1a) regardless of cell density. This indicated that the differing PM association of TRPV4 is due to trafficking changes, not expression level changes. GAPDH was used as a loading control. (D) Representative immunofluorescence (IF) images by confocal microscopy revealed TRPV4 (red) localization compared to the control protein transferrin receptor (TfR; green) in MCF10A, MCF10AT1, MCF10DCIS.com, and MCF10CA1a cells under ND and OC conditions. DAPI (blue) staining was used for visualizing the nuclei. As observed in the biochemical data in (B-C), cell crowding induces the relocalization of TRPV4 to the PM in MCF10DCIS.com cells. TRPV4 was associated with the PM in ND MCF10CA1a cells, with a clear elevated association in OC cells. Scale bar = 10 μm. (E) PM-associated TRPV4 (%) is quantified for the four cell lines under ND and OC conditions by line analysis, which showed a significant increase in both MCF10DCIS.com cells and MCF10CA1a cells due to cell crowding. The number of cells used for line analyses (technical replicates merged from three independent experimental repeats) was as follows: MCF10A (ND: 6; OC: 12), MCF10AT1 (ND: 6; OC: 11), MCF10DCIS.com (ND: 12; OC: 8), and MCF10CA1a (ND: 10; OC: 10). (F) IF images showed that hyperosmotic conditions induced by PEG 300 (74.4 mOsm/L) treatment also induced the relocalization of TRPV4 (red) to the PM in MCF10DCIS.com cells. TfR localization remained consistent under hyperosmotic conditions. Increased relocalization was also observed in MCF10CA1a cells. Scale bar = 10 μm. (G) The increased PM association of TRPV4 due to hyperosmotic stress was quantified by line analysis. The number of cells used for line analyses (technical replicates merged from two independent experimental repeats) was as follows: MCF10A (ND control: 6; ND +4% PEG: 15), MCF10AT1 (ND control: 6; ND +4% PEG: 9), MCF10DCIS.com (ND control: 12; ND +4% PEG: 8), and MCF10CA1a (ND control: 10; ND +4% PEG: 21). (H) Representative regions of interest (ROIs) of TRPV4-stained immunohistochemistry (IHC) images in different pathology phenotypes. High-grade ductal carcinoma in situ (DCIS) and invasive ductal cancer (IDC) ROIs clearly exhibit PM association of TRPV4. Two high-grade DCIS IHC images were acquired by two different people and both show PM-associated TRPV4. Scale bar = 20 μm. (I) Statistical results from independent histological evaluations of pathologies and TRPV4 distributions of 97 ROIs from 39 patient specimens indicated a high correlation (>70%) of PM association of TRPV4 with high-grade DCIS or IDC pathologies. Y/N: Yes/no, indicating both pathologists agreed that PM ion channels were present/absent. E: Equivocal, indicating the pathologists disagreed. Significantly high proportions of high-grade DCIS (75%) and IDC (73%) ROIs exhibited PM TRPV4 association, which was not observed in lower-risk cases. ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05, ns: p>0.05.
Image taken from Bu, et al. (2025). https://doi.org/10.7554/eLife.100490.4. Licensed under Creative Commons Attribution 4.0 International (CC BY 4.0).
Therefore, in this epithelial context, TRPV4 acts as a mechanical brake, where crowding actually promotes invasion by disabling mechanosensing. Critically, the plasma membrane localization of TRPV4, not its total expression, tracked with the observed phenotype. Membrane-associated TRPV4 inversely correlated with cell volume, frequency of Ca²⁺ transients, as well as invasion in vitro, and distinguished high-grade DCIS in patient tissue with high specificity relative to low-grade DCIS and normal ducts.
The study positioned TRPV4 not merely as a passive stretch sensor, but as a mechanically regulated signaling switch that translates physical compression and tissue stiffness into pro-invasive cellular behaviors in a context-dependent manner.
Thus, in breast cancer cells, TRPV4 acts as a mechanosensitive regulator that converts tumor crowding into altered calcium dynamics and increased invasiveness.
In the Tumor Stroma, TRPV4 Sustains Force-driven Metastatic Signaling
In pancreatic ductal adenocarcinoma (PDAC), the same channel produces the opposite outcome. Initial TRPV4 distribution in PDAC tissue was determined with Anti-TRPV4-ATTO Fluor-550 Antibody (#ACC-034-AO) (Figure 2) and here TRPV4 activity supported tumor progression through stromal mechanotransduction rather than in cancer cells directly (2). Expression of another mechanosensitive ion channel, Piezo1, was also assessed in the same cells using Anti-Piezo1 (extracellular) Antibody (#APC-087).
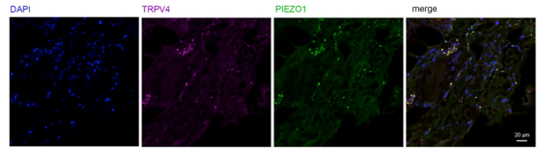
Figure 2. Immunostaining of human PDAC tissue sections with PIEZO1 and TRPV4 antibodies. Formalin-fixed PDAC tissue was stained with TRPV4-ATTO-Fluor-550 antibody at a dilution of 1:100 (magenta) or with PIEZO1 antibody at a dilution of 1:100 (green). DAPI was used to visualize the nuclei.
Image taken from Romac, et al. (2025). https://doi.org/10.1172/jci.insight.196280. Licensed under Creative Commons Attribution 4.0 International (CC BY 4.0).
Genetic deletion of TRPV4 reduced orthotopic tumor growth, diminished inflammatory cancer-associated fibroblast (CAF) marker expression, lowered systemic inflammatory cytokine levels Mechanistically, TRPV4 functioned downstream of mechanical sensing pathways in pancreatic stellate cells. Loss of TRPV4 blunted force-induced signaling programs that normally drive iCAF differentiation, inflammatory cytokine secretion, and long-range communication between the primary tumor and distant organs. Pharmacologic TRPV4 inhibition reproduced many of these systemic effects – including reduced circulating cytokines and liver niche signals – even when the effects on the primary tumor mass were modest at the tested dose.
Therefore, in a force-transmitting stromal setting, TRPV4 activity becomes permissive for metastasis.
In Cancer Cells, TRPV4 Links Stiffness Sensing to Growth and Therapeutic Response
In bladder cancer cells, TRPV4 again operates cell-autonomously, but now functions as a driver of aggressive behavior (3). Figure 3 shows that bladder cancer cells exhibited higher TRPV4 activity than normal urothelial cells as assessed via WB using Alomone’s Anti-TRPV4 Antibody (#ACC-034). In addition, TRPV4-dependent Ca²⁺ influx supported proliferation, transwell migration, cytoskeletal organization, focal adhesion assembly, matrix stiffness-dependent spreading, and responsiveness to mechanical cues across a range of substrate rigidities. Pharmacologic antagonism of TRPV4 reduced Ca²⁺ entry, suppressed proliferation, impaired migration, and disrupted cytoskeletal alignment in cancer cells while sparing normal urothelial cells.
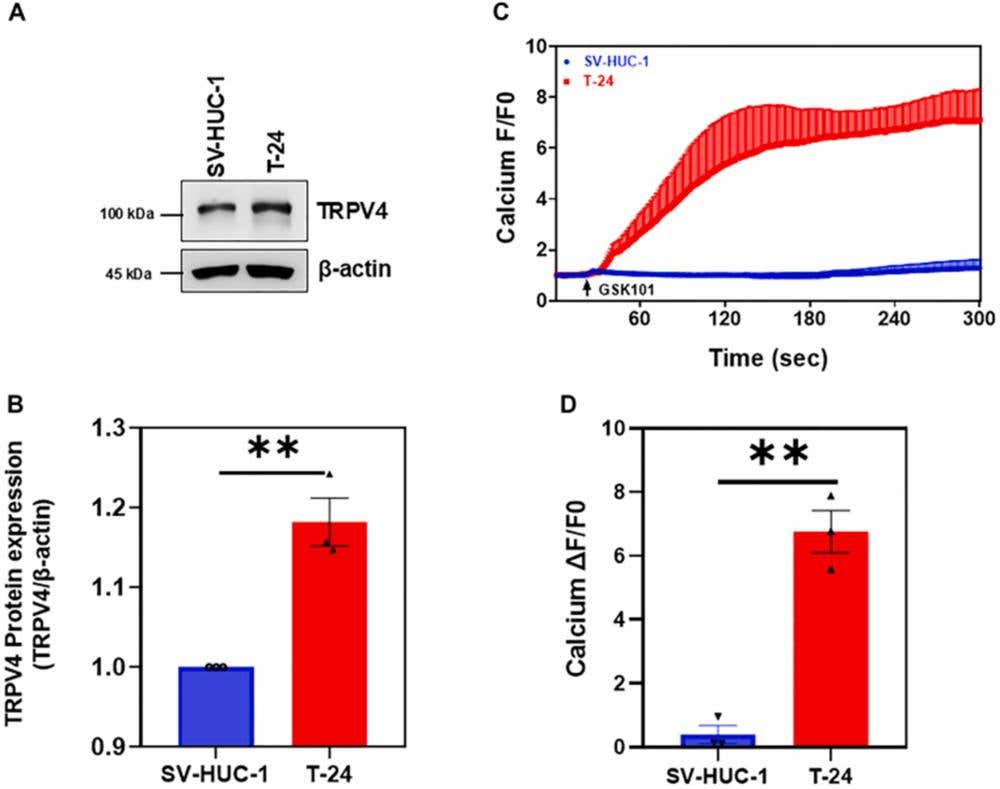
Figure 3. TRPV4 functional expression in bladder cancer cells. (A) Western blot (WB) showing TRPV4 expression in SV-HUC-1 and T-24 BLCA cells. TRPV4 protein expression was increased in T-24 BLCA cells compared to normal cells (SV-HUC-1), which correlated with increased calcium influx. (B) Quantification of WB analysis of TRPV4 normalized to β-actin. (C) Transients showing calcium influx in response to the TRPV4 agonist GSK1016709A (GSK101) in Fluo-4 loaded T-24 BLCA and SV-HUC-1 cells. (D) Quantification of values form C showed higher TRPV4-mediated calcium influx in T-24 BCLA cells than in SV-HUC-1 cells. The data are represented as the mean ± SEM from 3 independent experiments (n = 3). The statistical significance was determined using a Student’s paired t-test (∗∗P ≤ .01).
Image taken from Katari, et al. (2025). https://doi.org/10.1016/j.jpet.2025.103665. Licensed under Creative Commons Attribution 4.0 International (CC BY 4.0).
Notably, cisplatin-sensitive cells downregulated TRPV4 protein and activity after treatment, whereas chemoresistant cells retained TRPV4 expression, Ca²⁺ signaling capacity, and migratory behavior. In vivo, cisplatin treatment reduced TRPV4 levels in responsive xenografts, linking persistent TRPV4 signaling with therapeutic resistance.
Thus, in this context, the persistence of TRPV4 signaling tracks with mechanical adaptability, invasive behavior, and resistance to chemotherapy-associated stress.
A Unifying Mechanism
Taken together, these studies resolved a long-standing ambiguity regarding the role of TRPV4 in cancer. TRPV4 is not inherently oncogenic nor tumor suppressive – instead, it’s a mechanical state interpreter. In crowded epithelia, TRPV4 activity preserves cell volume, maintains Ca²⁺ transients, stabilizes epithelial mechanics, and suppresses invasion. In mechanically active tumor stroma, TRPV4 sustains force-dependent inflammatory signaling, cancer-associated fibroblast (CAF) differentiation, systemic cytokine release, pre-metastatic niche formation, and metastatic colonization. In stiff, force-responsive cancer cells, TRPV4 amplifies Ca²⁺-dependent programs that support proliferation, migration, cytoskeletal remodeling, and persistence under chemotherapeutic stress.
The same channel, coupled to different mechanical inputs and wired to distinct downstream programs, produces opposing phenotypes. These studies repositioned TRPV4 as a context-dependent mechanotransduction hub, where intervention or biomarker value depend on defining:
- the mechanical regime (crowding, stiffness, force transmission);
- the responding cell type (epithelial, stromal, tumor);
- the downstream Ca²⁺-dependent program engaged; and
- the functional state of the channel (localization, activity, persistence under stress).
Across models, the strongest signals come from functional readouts – membrane localization, Ca²⁺ flux, volume regulation, and resistance-associated persistence – not just from expression. Cancer progression is shaped by physical forces, but these forces do not act directly; instead, they require interpreters. In these studies, TRPV4 repeatedly serves as that interpreter, converting mechanical information into specific, measurable cancer phenotypes. The consequence is an interesting framework for the role of TRPV4 in cancer, one that helps define the mechanics, identify the signaling state, and the phenotype that follows. That principle, rather than channel activation per se, is what unifies these findings.


 Antibody")

 Antibody")
