Many ion channels are located at the nociceptor peripheral terminal, affecting neuron excitability after injury and as a result affecting pain sensation11. Voltage-gated Na+ and Ca2+ channels, TRP, ASIC, ligand-gated ion channels, P2X, NMDA, AMPA and Kainate receptors are some of the ion channels involved in the pathogenesis of pain10, several of which will be reviewed here.
Introduction
Pain is officially defined as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, as described in terms of such damage” by the International Association for the Study of Pain1. Actually, pain is a self-protective mechanism. The sense of pain occurs as the brain’s response to electrical and chemical changes that appear as a result of injury, disease or damage to the body2.
Pain is sensed and transmitted to the CNS by dorsal root ganglia (DRG) neurons which can be functionally divided into three subcellular compartments: 1. the nociceptor peripheral terminal which detects the pain causing stimuli (mechanosensation, thermal sensation and nociception)2,3. 2. the axon which conducts the nociceptive signal 3. the pre-synaptic terminal which transmits the signal forward to the next neuron and up to the brain where the signal is interpreted as pain (Fig. 1). Cell bodies of the sensory neurons that innervate the periphery are located at the trigeminal and dorsal root ganglia2. There are four major types of primary sensory neurons which can be differentiated morphologically and functionally: Aβ, Aα, Aδ and C fibers (Fig. 2)4-6.
There are two basic types of pain: neuropathic (nerve) pain and nociceptive (tissue) pain.
Neuropathic pain has been described in about 1% of the population. It is caused by a primary lesion or dysfunction in the nervous system. It can be subdivided into peripheral and central neuropathic pain depending on where the lesion or dysfunction has occurred. Peripheral neuropathic pain may result from disease, while central neuropathic pain is caused by damage to the spinal cord or the brain1. Neuropathic pain lasts long after apparent healing of the damaged tissues and frequently it turns into chronic pain which serves little if any protective effect. Neuropathic pain actually interferes with Contribution of Ion Channels in Pain Sensation the restoration of normal function after injury or disease2,7,8.
Nociceptive pain can be subdivided into somatic pain (cutaneous or deep tissues) and visceral pain (pain in the internal organs). Nociceptive pain occurs when nociceptors are stimulated in response to substances that are released from damaged or inflamed tissue. This combination of substances is called the ‘inflammatory soup’ and it includes extracellular protons, nucleotides, nerve growth factor, serotonin, bradykinin, and others2. The nociceptor peripheral terminals can also respond to mechanical and thermal stimuli (Fig.1). Nociceptive pain can be sharp, dull or aching. It is usually time limited (diminishes once the tissue damage heals) and serves a protective purpose2,7,8.
All three stages of the pain pathway, described above, involve ion channels and receptors which are located at the nociceptor peripheral terminals and are the first to detect the pain causing stimuli (heat, pressure, tissue damage etc.) and transmit them further. Sometimes, modulation of ion channel activity by components of the inflammatory soup may lead to pathological states such as allodynia (pain resulting from a stimulus that is normally not painful, such as light stroking of the skin which under normal conditions is not painful, but can be excruciating to sunburned skin) and hyperalgesia (an excessive response to noxious stimuli)2,3,9. To date, ion channels have become an important target in the search for new pain therapies10.
Role of Ion Channels in Pain
The term ‘ion channels’ includes members of multiple gene families, which are responsive to various stimuli. Many of these ion channels are located at the nociceptor peripheral terminal, affecting neuron excitability after injury and as a result affecting pain sensation11. Voltage gated Na+ and Ca2+ channels, TRP, ASIC, ligand gated ion channels, P2X, NMDA, AMPA and Kainate receptors are some of the ion channels involved in the pathogenesis of pain10, several of which will be reviewed here.
1. Detection of Pain
Transient Receptor Potential Channel (TPRP)
The mammalian sensory system can feel a wide range of temperatures12-13. Temperatures below 15°C or above 43°C evoke thermal sensation accompanied by the sensation of pain. Six thermosensitive ion channels have been described, all members of the TRP family. Each channel exhibits distinct thermal activation thresholds; TRPV4 (>25°C), TRPV3 (>31°C), TRPV1 (>43°C), TRPV2 (>52°C), TRPM8 (<28°C) and TRPA1 (<17°C)12. The involvement of the TRPV1 channel (also known as the capsaicin receptor or vanilloid receptor, VR1) in thermal nociception has been well documented by different methods12. TRPV1 is expressed predominantly in nociceptors and in sensory neurons and is suggested to be associated with tissue injury and infl ammation. TRPV1 is activated by heat and capsaicine12-15. The inflammatory mediators, ATP and bradykinin (figure 3), were reported to potentiate TRPV1 through the P2Y2 and B2 receptors, respectively, in native DRG neurons and in a heterologous system12-14.
It appears that TRPV1 is upregulated in inflammatory conditions, thus it is considered to be an integrator of inflammatory pain pathways12. In humans, changes in TRPV1 expression are connected to inflammatory bowel disease and to irritable bowel syndrome15. The other members of the thermosensitive ion channel, TRPV2, TRPM8 and TRPA1 have activation thresholds within the noxious range of temperatures indicating possible involvement in thermal nociception12.
P2X Receptors
Another important and well-documented player in pain processing is the P2X3 ion channel. The P2X3 channel belongs to the ligand gated ion channel family that consists of seven receptor subtypes named P2X1-P2X7 and is activated by extracellular ATP16,17. ATP is the primary source of energy for a variety of cellular reactions such as muscle contractions, protein synthesis etc18. However, recent data implicates a new and very different role for ATP: activator of pain receptors. ATP released from damaged or inflamed cells activates the P2X3 receptor to initiate nociceptive signals18,19. The P2X3 receptor is highly expressed on nociceptive sensory neurons in DRG both as a homomer and as a heteromer (P2X3/P2X2)20. Upregulation of P2X3 receptor in dorsal horn as well as in DRG was reported following neuropathic injury in animal models18,21. A reduction in injury and chronic nociceptive pain was demonstrated using a selective, non-nucleotide antagonist (A-317491) to the P2X3 receptor in both its homomeric and heteromeric forms22. Although P2X3 and P2X3/2 do not seem to have a major role in mediating acute inflammation or visceral pain they are still a possible target for the development of pain killers.
Acid Sensing Ion Channels (ASICs)
Tissue acidosis is associated with inflammation and is an significant source of pain23,24. During inflammation, the extracellular pH values decrease (below pH=6), activating nociceptors by gating the ASICs24. The ASICs are sodium channels belonging to the degenerin/ENaC superfamily. Six isoforms have been identified encoded by four different genes: ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3 and ASIC425. The most interesting, in the context of pain, is ASIC3, which is predominantly expressed in DRG neurons making it a good candidate for a pain sensor. In humans ASIC3, contributes to hyperalgesia and allodinia in inflammation. ASIC3 is more widely distributed in humans than in mice, which might indicate a more extensive role in human nociception.
2. Propagation of the Pain Signal
Voltage Gated Na+ Channels
Voltage gated Na+ channels (VGSCs, NaV) are key players in electrical conduction along axons, including those of primary sensory neurons. They are thought to play a crucial role in several chronic painful neuropathies resulting from injury to the peripheral nerves26. Physiological as well as pharmacological evidence suggest that VGSCs play a major role in the development and the maintenance of hyperexcitability observed in primary afferent neurons as a result of nerve or tissue injury27.
The voltage gated Na+ channels are a family of nine structurally related α subunits (NaV1.1 to NaV1.9) which show distinct expression patterns and are associated with one or more accessory β subunits (β1 to β3)26,28-30. The expression of the α subunit isoforms is developmentally regulated and tissue specific. VGSC are classified into two groups according to their sensitivity to Tetrodotoxin (TTX): TTX-sensitive and TTX-resistant channels26, 31. With the exception of NaV1.4, it seems that all VGSCs are expressed to some extent in sensory neurons26,27,32.
TTX-sensitive VGSCs
Nav1.6 is present in most sensory neurons. Nav1.3 is highly expressed in embryonic sensory neurons but its level dramatically decreases in adult sensory neurons27. Recently, data describing up regulation of expression of the Nav1.3 channel in injured neurons and injured spinal cord was reported33, 34. Nav1.7 is predominantly expressed in dorsal root ganglion (DRG). Dominant mutations in the Nav1.7 gene are associated with erythermalgia (a rare autosomal disease characterized by sporadic burning pain accompanied by redness and heat in the extremities). These data might confirm a possible and important role for Nav1.7 in nociception35-37.
TTX-resistant VGSCs
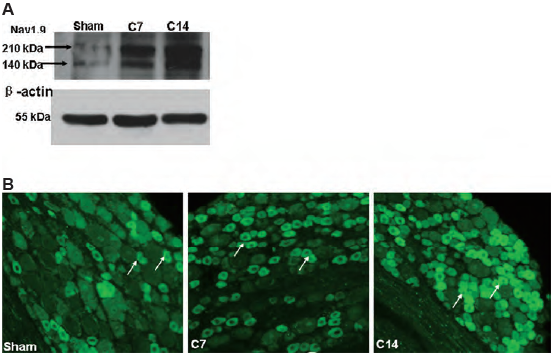
Of special interest are the two TTX-resistant channels, NaV1.8 and NaV1.9 which are expressed predominantly in small diameter DRG. NaV1.9 is expressed in C-fibers while NaV1.8 is expressed in A fibers27. To date, little is known about the role that NaV1.9 plays in neuropathic pain. So far the accumulated data does not suggest any contribution despite the location of the channel in DRG27. The kinetic properties of NaV1.9 along with computer analysis suggest that NaV1.9 is active at the resting membrane potential and might modulate the resting potential of nociceptors and their response to sub-threshold stimuli26,38. Although NaV1.9 is believed to be expressed only in small diameter DRG, recent data have demonstrated its location in the hippocampus where it is activated by BDNF. The findings that NaV1.9 expression is not restricted to DRG alone make it much less attractive candidate for pain therapeutic26. In contrast, inhibition of NaV1.8 channels has been found effective in reducing pain following injury to somatic afferent axons or tissue inflammation. It has also been demonstrated that NaV1.8 channels are involved in the activation of afferent nerves after chemical irritation of the bladder39-40.
Drugs directed against NaV1.8 are already under development by IONIX Pharmaceuticals based on NaV1.8 structure and function. One is a NaV1.8 blocker and the other is NaV1.8 regulator which down-regulates functional expression of NaV1.8 channels41.
The role of NaV1.8 and NaV1.9 in pain is still controversial. Several points support their involvement in the pain process:
- The predominant expression within the DRG neurons of a number of Na+ channels.
- The change of pattern and level of expression following injury or inflammation.
However, the mechanism by which Na+ channels affect pain sensation still remains to be elucidated.
3. Transmission of the Pain Signal to the CNS
Voltage Gated Ca2+ Channels
Voltage gated Ca2+ channels (VGCCs) are also expressed in nociceptors, predominantly at the DRG neuron presynaptic terminals in the dorsal horn where they control the release of neurotransmitters42. The Ca2+channel is composed of several subunits, the pore forming α1 subunit and the auxiliary subunits; β, γ, α2δ which modulate α1 function. The α1 subunit defines the channel subtype. To date, ten genes encoding VGCCs have been identified and grouped into three families: CaV1. family (CaV1.1- 1.4 correspond to L-type current), the CaV2. family which includes CaV2.1 (corresponding to P/Q current), CaV2.2 (N-type current), CaV2.3 (R-type current) and the CaV3. (CaV3.1-3.3 correspond to the T-type current)42.
N-type Ca2+ Channels
In the search for painkillers or pain relievers, special interest has been directed towards the N-type channel, CaV2.2. Several lines of evidence implicate N-type Ca2+ channels as critical for the transmission of pain signals at the spinal cord level43. Release of neuropeptides such as substance P was stopped by the use of N-type Ca2+ channel blockers. Inhibition of these channels led to suppression of pain42. Decrease in sensitivity to neuropathic and inflammatory pain was demonstrated in knockout mice lacking the gene encoding the N-type channel42. The CaV2.2 channels that are expressed on sensory neurons are the target for morphine (via GPCR opioid receptor) and other N-type selective peptide blockers that were shown to cause relief from pain when injected into or around the spinal cord, both in humans and animals44. However, those peptides might also cause severe side effects due to non selective blockade of all N-type channels and not specifically to those located on nociceptors. Recently, an alternative spliced isoform of the N-type channel was found to be expressed primarily on a subset of small nociceptive neurons44,45. Drugs directed against this isoform may not have side effects, since they block a specific channel. Several small organic blockers of the N-type channel are already under development despite their side effects.
P/Q type Ca2+ Channels
Migraine is characterized by severe attacks of unilateral headache which troubles 10-15% of the population. Missense mutations in the Ca2+ channel CaV2.1 (P/Q-type) have been found to be the cause of migraine in about 50% of tested families suffering from familial hemiplegic migraine46-48.
T-type Ca2+ Channels
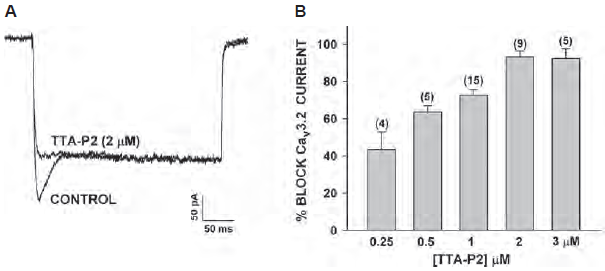
T-type Ca2+ channels were first described in the peripheral sensory neurons of the DRG, however their function is still unclear42. Although there is evidence implicating a role in pain processing the lack of selective blockers for T-type channels has made it difficult to assess their role in manifestations of neuropathic pain49.
The α2δ-1 Auxiliary Subunit
There is evidence implicating an important role for the auxiliary subunit α2δ-1 in neuropathic pain processing. The α2δ-1 subunit in the DRG is upregulated, at both the mRNA and protein levels, following experimental nerve-injury50. It binds gabapentin (an anticonvulsive drug effective against clinical neuropathic pain) with high affinity. Gabapentin injected intrathecally suppressed allodynia in a dose dependent manner in an animal model without any effect on behavior of nonallodynic rats51. Since α2δ-1 upregulation was not accompanied by upregulation of the α1 subunit nor does the β auxiliary subunit it seems that it have a distinct functional role in addition to the modulation of VGCCs51.
Conclusion
Data is accumulating indicating a crucial role for ion channels in pain pathways, and many ion channels have already become candidates for therapeutics and drug development. However, the mechanisms of actions have not been fully elucidated, and the issues of specificity and side effects are still remaining to be dealt with.
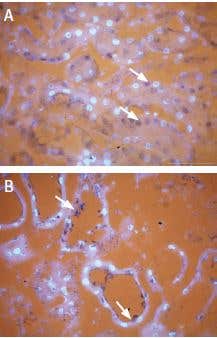
TRPV1 expression in kidney. Staining of TRPV1 in kidney using Anti-TRPV1 (VR1) Antibody (#ACC-030). Selective subsets of cells were stained in mouse (A) compared to rat (B). Diaminobenzidine color product is blue-black (arrows). The fluorescent counterstain is DAPI (silver).

Figure 1. Pain Signal Pathway.
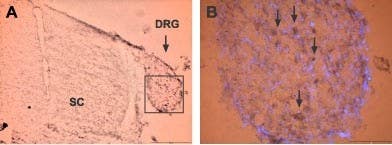
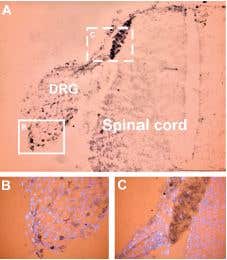
ASIC3 expression in DRG. Staining of ASIC3 with Anti-ASIC3 Antibody (#ASC-018) in rat embryo dorsal root ganglion. A) Low power magnification shows the distribution of staining in DRG and spinal cord. B) Higher power magnification shows distribution of ASIC3 immunoreactive cells (arrows) within DRG (box in A is enlarged in B). The color product is brown-black (diaminobenzidine). The counterstain is the fluorescent dye DAPI (blue).
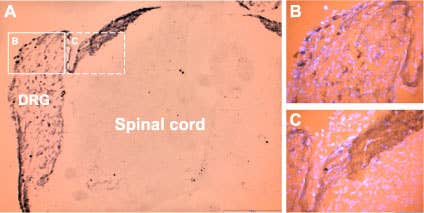
P2X3 expression in DRG. Staining of P2X3 in rat dorsal root ganglion (DRG) with Anti-P2X3 Antibody (#APR-016). Cells within the DRG were stained (see solid line frame enlarged in B) as well as fibers and the area of entry of dorsal root into spinal cord (see dashed line frame enlarged in C). The counterstain in B and C is DAPI, a fluorescent dye visualized in the UV range.
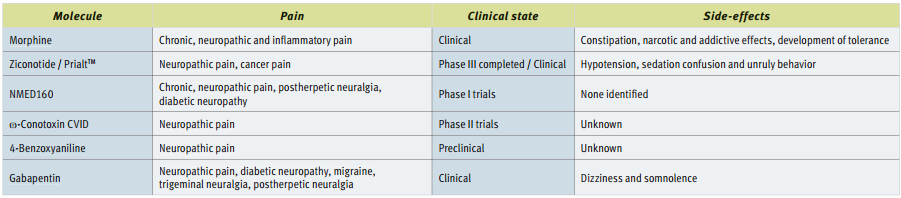
N-Type Blockers in Use and Under Development.43
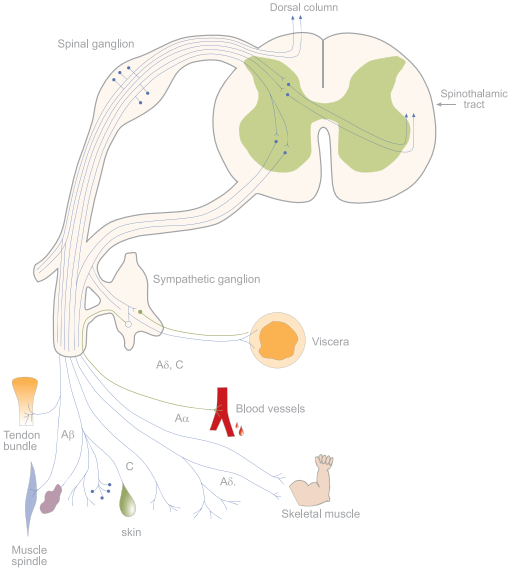
Figure 2. Sensory Neurons Types.
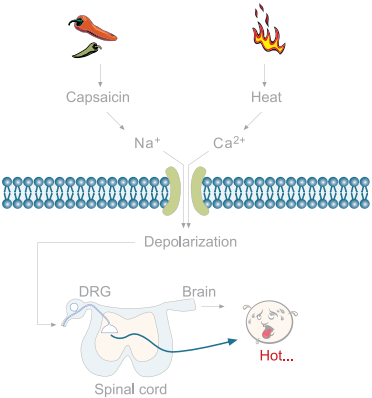
Figure 3. The Activation of TRPV1.
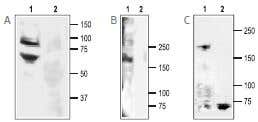
Western blotting of rat DRG lysate:
A. 1. Anti-ASIC3 Antibody (#ASC-018) (1:200).
2. Anti-ASIC3 Antibody, with the negative control antigen.
B. 1. Anti-SCN11A (NaV1.9) Antibody (#ASC-017) (1:200).
2. Anti-SCN11A (NaV1.9) Antibody, with the negative control antigen.
C. 1. Anti-NaV1.8 (SCN10A) Antibody (#ASC-016) (1:200).
2. Anti-NaV1.8 (SCN10A) Antibody, with the negative control antigen.