Voltage-gated Na+ channels are membrane proteins that are essential for the generation and propagation of action potentials in excitable tissues, such as brain, muscle, and heart following membrane depolarization. These channels are heteromultimeric protein complexes consisting of one α and one or two β subunits. Alomone Labs offers an extensive list of primary polyclonal antibodies as well as modulators related to this family. The use of Alomone Labs antibodies in fluorescence immunocytochemistry and western blot analyses demonstrates membrane expression of the different subtypes of voltage-gated Na+ channels and allow to monitor the developmental and physiological changes as described and presented in the article below.
Introduction
There are nine recognized members of the voltage-gated Na+ channel family (VGSC; NaV1.1-NaV1.9). Of these, NaV1.1, NaV1.2, NaV1.3 and NaV1.6 are highly (but not exclusively) expressed in the central nervous system (CNS), whereas NaV1.7, NaV1.8 and NaV1.9 demonstrate a more restricted expression pattern in autonomic and sensory neurons of the peripheral nervous system (PNS). NaV1.4 and NaV1.5 represent the predominant skeletal muscle and cardiac Na+ channels, respectively. The NaV1.6 subtype is also highly expressed in the peripheral nervous system where it is enriched at the nodes of Ranvier of myelinated axons and contributes to saltatory conduction4,18.
The expression of the different subtypes of voltage-gated Na+ channels is variable within different types of neurons, or within different parts of the membrane of a single neuron. The subtype composition, density and distribution of voltage-gated Na+ channels determine the property of the various types of neurons or the specific areas of each neuron26. Also, expression and distribution of the different members of the voltage-gated Na+channel family at the subcellular level, differs during the development19.
The expression of voltage-gated Na+ channels is a dynamic process, and alterations in the physiological state as well as injury induce changes in their expression, which can alter neuronal behavior.
Localization and Distribution of Voltage-Gated Na+ Channels
A study of the normal distribution of voltage-gated Na+ channels using immunohistochemistry and immunolabelling analyses showed that NaV1.3 (using Anti-SCN3A (NaV1.3) Antibody (#ASC-004)), and other voltage-gated Na+ channels are expressed in the human optic nerve1.
The distribution of voltage-gated Na+ channels in spiral ganglion neurons (SGN) was examined using RT-PCR and immunohistochemistry analyses. The use of Anti-SCN1A (NaV1.1) Antibody (#ASC-001), Anti-NaV1.6 (SCN8A) Antibody (#ASC-009), and Anti-NaV1.7 (SCN9A) Antibody (#ASC-008) demonstrated that the respective channels are localized in SGN cell bodies and in axonal processes11.
A study compared voltage-gated Na+ channels in cerebellar Purkinje cells from mormyrid ish and rat using immunohistochemical and electrophysiological methods. Results show that NaV1.1, NaV1.2, and NaV1.6 channels are present in comparable densities and locations in the mormyrid and rat cerebellum, using their respective Alomone Labs antibodies, and also share the same set of Na+ conductance8.
Axon initial segments (AISs) and nodes of Ranvier contain high densities of voltage-gated Na+ channels. AISs are a structurally and functionally specialized region of the axon believed to be critical for the generation of action potentials (AP). The high density of voltage-gated Na+ channels at AISs20 lowers the threshold for AP initiation and gives rise to a fast, regenerative inward current during the rising phase of the depolarization. The nodes of Ranvier ensure efficient AP conduction.
Morphologically differentiated cerebellar granule (CG) cells express NaV1.2 and NaV1.6, though both subunits appear to be differentially regulated. Immunocytochemical analysis, using Alomone Labs antibodies showed that NaV1.2 is localized at most AISs of CG cells from 8 days in vitro (DIV 8 to DIV 15). At DIV 8, NaV1.6 was found uniformly throughout the somata, dendrites and axons with occasional clustering in a subset of AISs. Accumulation of NaV1.6 at most AISs was evident by DIV 13–14 (Figure 1), suggesting it is developmentally regulated at AISs22.

CG cells were labeled with Anti-NaV1.6 (SCN8A) Antibody (#ASC-009) at DIV 8 (A) or DIV 12 (B). Right columns show immunoreactivity for NaV1.6 when the primary antibody was preincubated with the immunizing peptide. At DIV 8, NaV1.6 staining was diffusely distributed and rarely concentrated at the AIS whereas accumulation was detected in most AISs at DIV 12.
Adapted from reference 22 with permission of Blackwell Publishing Ltd.
Nodes of Ranvier in myelinated axons in the CNS are ideal places to examine how certain subtypes of voltage-gated Na+ channels are inserted into a specific region of a neuron. During development, NaV1.2 first appears in the predicted nodes during myelination, and NaV1.6 replaces it in the mature nodes25,26. These characteristic localizations are influenced by myelination. Examination of the influence of the paranodal junction on the switching of Na+ channel subunits was done using the sulfatide-deficient mouse model (this mutant displays disruption of paranodal axoglial junctions). The initial switching of NaV1.2 to NaV1.6 in the optic nerve was observed using Anti-NaV1.6 (SCN8A) Antibody. However, the number of NaV1.2-positive clusters was significantly higher than in wild-type mice suggesting that paranodal junction formation may be necessary for complete replacement of nodal NaV1.2 to NaV1.6 during development as well as maintenance of NaV1.6 clusters at the nodes26.
NaV1.2 and NaV1.6 are selectively targeted to the proximal and distal AISs, respectively (in the rat prefrontal cortex)13, in AISs of CA1 hippocampal pyramidal neurons23 and in mature Purkinje neuron initial segments14. NaV1.6 channels accumulate at the distal AIS to determine the lowest threshold for AP initiation. On the other hand NaV1.2 channels are highly clustered at the proximal AIS guaranteeing that the AP initiated at the distal AIS will not back-propagate into the soma as shown in immunohistochemical studies in rat prefrontal cortex using Anti-SCN2A (NaV1.2) Antibody and Anti-NaV1.6 (SCN8A) Antibody as well as Anti-Pan NaV Antibody (#ASC-003)13 (Figure 2).
A) Antibody staining for AnkG (red) and NaV1.2 with Anti-SCN2A (NaV1.2) Antibody (#ASC-002), (green) in the rat prefrontal cortex. Note that the proximal AIS has strong staining for NaV1.2. B) Double staining for AnkG and NaV1.6 with Anti-NaV1.6 (SCN8A) Antibody (#ASC-009), (green). Note that the distal region of the AIS is heavily stained. C) Double staining for AnkG and NaV with Anti-Pan NaV Antibody (#ASC-003), (green). Plots of the averaged (± s.e.m.) fluorescence intensity as a function of distance from the soma at the AIS are shown. Images are projections of confocal z stacks. Scale bars 10 μm. Error bars represent s.e.m.
Adapted from reference 13 with permission of Macmillan Publishers Ltd.
In different types of neurons the expression of various Na+ and K+ channel subunits varies along the proximo-distal axis of AIS. This precise arrangement is likely to contribute to the diversity of firing properties observed among central neurons20. Using an antigen retrieval method, Anti-NaV1.6 (SCN8A) Antibody was used to monitor the distribution of NaV1.6 which was found to be expressed in AISs of different neurons in the neocortex, hippocampus, main olfactory bulb (MOB) and cerebellum along with other channel subunits20.
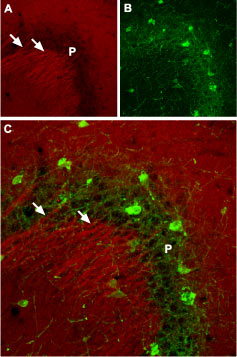
Another direction of research was done to investigate the mechanism for targeting and restricting voltage-gated Na+ channels to excitable membranes. It was proposed that one or more proteins colocalize with Na+ channels including βIV spectrin, ankyrin-G (ankyrin-3), and the L1 cell adhesion molecule (L1 CAM)14,15. Immunohistochemistry studies showed that ankyrin-G and IV spectrin appear at AIS by P2, whereas L1 CAM and NaV1.6 (using Alomone Labs’ antibody) are not fully assembled at continuous high density along AISs until P9. Furthermore,NaV1.6 and L1 CAM are not clustered in adult Purkinje neuron initial segments of mice lacking cerebellar ankyrin-G. These results support the conclusion that ankyrin-G coordinates the physiological assembly of voltage-gated Na+ channels, in AISs14. Similarities were found in the clustering of ankyrin-G, defining earlydevelopmental intermediates in the nodes of Ranvier formation in the mouse optic nerve14 (Figure 3).

Sections of P9 rat cerebellum were double labeled with antibodies against=ankyrin-G (green), and NaV1.6 using Anti-NaV1.6 (SCN8A) Antibody (#ASC-009), (red). Arrowheads indicate Purkinje cell initial segments. Scale Bars, 10 μm.
Adapted from reference 14 with permission of Rockefeller University Press.
Contactin (also known as F3, F11 in various species) is a glycosyl-phosphatidylinositol (GPI)-anchored protein, expressed in neurons and glia. In the CNS, there is a high level of colocalization of contactin and voltage-gated Na+ channels at the nodes of Ranvier, both during development and in the adult. A combination of biochemical, electrophysiological, and immunolocalization experiments (in optic nerves of rat, using Anti-Pan NaV Antibody) all pointed to a specific association of contactin with voltage-gated Na+ channels and concluded that contactin may influence the functional expression and distribution of Na+ channels in neurons16.
In another study, in order to understand the mechanisms and sequences responsible for targeting and localizing NaV1.2 tounmyelinatedaxons, chimeras between NaV1.2 and NaV1.6 were generated (NaV1.6 does not localize at unmyelinated axons). Results suggest that the 451 amino acids of NaV1.2 C-terminal are likely required for its interaction with neuron-specific factors in order to direct it to axons as tested using Anti-SCN2A (NaV1.2) Antibody19.
Neuronal Disorders Involving Voltage-Gated Na+ Channels
Multiple Sclerosis/Myelination
The optic nerve axon is a CNS tract commonly affected in multiple sclerosis (MS). Numerous studies investigating the organization of Na+ channels along the axons of the optic nerve, were carried in experimental allergic encephalomyelitis (EAE) mice, a model of MS. Immunocytochemical analysis using Anti-SCN2A (NaV1.2) Antibody and Anti-NaV1.6 (SCN8A) Antibody, demonstrated a significant switch from NaV1.6 to NaV1.2 expression in the optic nerve in EAE. In addition, there was a reduction in frequency of NaV1.6-positive nodes and increased frequency of NaV1.2-positive nodes. These findings suggest that electrogenesis in EAE may revert to a stage similar to that observed in immature retinal ganglion cells in which NaV1.2 channels support conduction of action potentials along axons6.
The expression of NaV1.2 and NaV1.6, Na+/Ca2+ exchanger (NCX) and β-amyloid precursor protein (β-APP a marker of axonal injury), were examined in the spinal cord dorsal columns of mice with (EAE)5 and in postmortem cervical spinal cord and optic nerve tissue, from patients with disabling secondary progressive MS7. Using triple labeled fluorescent immunohistochemistry, it was shown that NaV1.6 and NCX are colocalized with β-APP in acute MS lesions. The result demonstrate the molecular identities of the Na+ channels expressed along demyelinated and degenerating axons in MS and suggest that the coexpression of NaV1.6 and NCX is associated with axonal degeneration in MS5,7. Furthermore, there was a significant increase in the number of demyelinated axons demonstrating diffused NaV1.6 and NaV1.2 channels in EAE using Anti-SCN2A (NaV1.2) Antibody and Anti-NaV1.6 (SCN8A) Antibody5 (Figure 4).

A) Histogram demonstrating the percentage of β-APP-positive axons that are NaV1.6-positive, NaV1.6/NaV1.2-positive (i.e., co-express both NaV1.6 and NaV1.2), or NaV1.2-positive in EAE. B) Digital images of a representative field demonstrating axonal profiles in EAE spinal cord immunostained for β-APP (blue; top panel), NaV1.6 and NaV1.2 using Anti-NaV1.6 (SCN8A) Antibody (#ASC-009), (red; middle panel) and Anti-SCN2A (NaV1.2) Antibody (#ASC-002), (green; bottom panel) respectively. Arrows point to β-APP-positive and NaV1.6-positive axons that do not display NaV1.2 immunostaining.
Adapted from reference 5 with permission of Oxford University Press.
Demyelination can be repaired by remyelination in both humans and rodents, and even within the CNS. Congenitally dysmyelinated spinal cord axons can undergo remyelination, by transplanting adult neural precursor cells (aNPCs) from the brain of transgenic mice into the spinal cords of adult Shiverer (shi/shi) mice, which lack compact CNS myelin. Six weeks after transplantation, the transplanted aNPCs expressed oligodendrocyte markers, and formed compact myelin10. Anti-NaV1.6 (SCN8A) Antibody and Anti-SCN2A (NaV1.2) Antibody were both used to show that Na+ channels are not affected by dysmyelination. Specifically, the lack of immunoreactivity is as expected and that of NaV1.6 remains clustered at the nodes of Ranvier10. Similarly, glial cells, from olfactory ensheathing cells were transplanted into the demyelinated spinal cord and subsequently formed a compact myelin. Immunocytochemical analysis in remyelinated axons, with antibodies purchased from Alomone Labs, showed that NaV1.6 was clustered at nodes of Ranvier, whereas KV1.2 was aggregated in juxtaparanodal regions, recapitulating the distribution of these channels within mature nodes of uninjured axons. These findings indicate that, in addition to forming myelin, glial cell transplantion provides an environment that supports the development and maturation of nodes of Ranvier and the restoration of impulse conduction in central demyelinated axons24.
Cerebro spinal fluid (CSF) from patients with Amyotrophic Lateral Sclerosis (ALS) evokes a change in ion channel expression in newborn rat spinal motor both in vivo and in vitro. Following exposure of CSF from ALS patients, cultures and spinal cord sections were processed for immunostaining of NaV1.6 using Anti-NaV1.6 (SCN8A) Antibody. A decrease in the expression of NaV1.6 in motor neurons in the ALS-CSF treated group was observed; thereby indicating that altered expression of NaV1.6 may interfere with the electrical activity of motor neurons, and thereby leads to the degeneration of neurons12 (Figure 5).

Neonatal rat spinal cord sections were labeled with ChAT (green, A), a marker for spinal motor neuron and NaV1.6 using Anti-NaV1.6 (SCN8A) Antibody (#ASC-009), (red, B). The merged image (C) confirms that NaV1.6 is expressed in motor neurons. Arrows indicate the spinal motor neurons in the ventral horn of spinal cord showing NaV1.6 labeling co-localised to ChAT positive neurons. Absence of staining in the negative control (D) confirms specificity of staining. Scale bars, 75 μm (A–D). Note a decreased NaV1.6 expression in rat pups injected with ALS-CSF (H), when compared to those injected with non ALS-CSF (G), sham control (F) and normal control (E).
Adapted from reference 12 with permission of Elsevier.
Ischemia and Nerve Injury
Alterations in the expression, molecular composition, and localization of voltage-gated Na+ channels play major roles in a broad range of neurological disorders. Alteration of Na+ channel function has been reported to occur in a variety of neuropathological states including axonal degeneration, epilepsy and brain injury. Changes in voltage-gated Na+ channel activity can play a role in reorganization, recovery, or possibly excitotoxic damage following injury.
Prolonged hypoxemia in fetal llama brain caused a significant decrease in the expression of NaV1.1 and less so for NaV1.2 channels detected with Anti-SCN1A (NaV1.1) Antibody and Anti-SCN2A (NaV1.2) Antibody, respectively9. The decrease in their expression was not accompanied by cell death suggesting that the fetal llama brain responded to hypoxemia with an adaptive hypometabolism as opposed to a more degenerative effect.
In order to gain further insight into the molecular events underlying brain injury, male rats were subjected to ischemic brain injury. Immunoreactive analysis of brain tissue, using Anti-SCN1A (NaV1.1) Antibody revealed a qualitative decrease in protein levels of NaV1.1 throughout the ischemic regions, beginning at the early stage of injury (6h) with dramatic losses at later stages (24 and 48h)28, (Figure 6).

NaV1.1 immunoreactive cells (dark brown) with Anti-SCN1A (NaV1.1) Antibody(#ASC-001) counterstained with cresyl violet (light purple) from striatal brain regions of sham animals and at 2, 6, 24 and 48 h post-MCAo. A) Contralateral; B) Ipsilateral. Sham images taken 24 h following sham surgery. Scale bars, 50 μm.
Adapted from reference 28 with permission of Elsevier.
Voltage-gated Na+ channel proteolysis occurs in an early event following ischemia and traumatic brain injury. A recent study showed that activated calpain causes voltage-gated Na+ channel proteolysis. Using Anti-Pan NaV Antibody and Anti-SCN2A (NaV1.2) Antibody, it was shown that calpain-mediated voltage-gated Na+ channel cleavage may play an important pathophysiological role in recovery or demise after injury27.
Neurogenesis in the neocortex following ischemia has been for long a controversial issue. Using retrovirus-mediated labeling of neocortical layer 1 proliferating cells with membrane-targeted green fluorescent protein (GFP), it was found that the neocortical layer is a source of adult neurogenesis under ischemic conditions. In addition, it was found that a large number of the GFP-positive cells express voltage-gated Na+ channels, which are essential for the production of action potentials, of which 98 of 162 GFP-positive cells were NaV1.6-positive (using Anti-NaV1.6 (SCN8A) Antibody), strongly suggesting that biogenesis not only occurs at the expression level but also at the functional level21.
Epilepsy
CNS plasticity is essential for normal function, but can also reinforce abnormal network behavior, leading to epilepsy and other disorders. Certain types of epilepsy can result from direct voltage-gated Na+ channel gene mutations or changes in genes that affect voltage-gated Na+ channel expression pathways, ultimately predisposing neurons toward repetitive firing and network hyperexcitability. Because voltage-gated Na+ channels can underlie neuronal bursting, it was asked whether there are differences in the expression of voltage-gated Na+ channels in the rodent model of absence epilepsy (WAG/Rij) cortex compared to non-epileptic controls. Immunocytochemistry analysis showed that protein levels of NaV1.1 and NaV1.6 were upregulated in layer II–IV in the cortical neurons17, using Alomone Labs specific antibodies (Figure 7). This region of the cortex approximately matches the electrophysiologically determined region of seizure onset17. Early treatment with ethosuximide blocks changes in the expression of NaV1.1 and NaV1.6, normally associated with epilepsy in this model, leading to the suppression of seizures even after therapy was discontinued. These findings suggest that early treatment during the development provides a new strategy for preventing epilepsy2.

Immunostaining in control rats (no *) versus WAG/Rij rats (*) using Anti-SCN1A (NaV1.1) Antibody (#ASC-001), (A, A*), Anti-SCN2A (NaV1.2) Antibody (#ASC-002), (B, B*), and Anti-NaV1.6 (SCN8A) Antibody (#ASC-009), (C, C*). The control and epileptic images were digitally contrast-enhanced using identical parameters to qualitatively illustrate the up-regulation of the Na+ channels. D) Optical intensity quantification from unenhanced images of individual neurons in layers II-IV shows increased staining with Anti-SCN1A (NaV1.1) Antibody and Anti-NaV1.6 (SCN8A) Antibody in the epileptic animals (V) compared to controls (V), indicating up-regulated Na+ channel expression.
Adapted from reference 17 with permission of Elsevier.
Kindling is a form of abnormal activity-dependent facilitation that can be initiated, developed, and expressed in wild-type mice. Voltage-gated Na+ channel expression by immunocytochemistry was verified using Alomone Labs-related antibodies. It was found that kindling is associated with increased expression of NaV1.6, which occurs selectively in hippocampal CA3 neurons, while no changes regarding the expression of NaV1.1 and NaV1.2 was observed. These results suggest an important role for altered expression of voltage-gated Na+ channels in the abnormally enhanced excitability seen in epilepsy and other chronic disorders of the nervous system3.
References
- Barron, M.J. et al. (2004) Br. J. Ophthalmol. 88, 286.
- Blumenfeld, H. et al. (2008) Epilepsia 49, 400.
- Blumenfeld, H. et al. (2009) Epilepsia 50, 44.
- Caldwell, J.H. et al. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5616.
- Craner, M.J. et al. (2004) Brain 127, 294.
- Craner, M.J. et al. (2003) Brain 126, 1552.
- Craner, M.J. et al. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 8168.
- de Ruiter, M.M. et al. (2006) J. Neurophysiol. 96, 378.
- Ebensperger, G. et al. (2005) J. Physiol. 567, 963.
- Eftekharpour, E. et al. (2007) J. Neurosci. 27, 3416.
- Fryatt, A.G. et al. (2009) Mol. Cell Neurosci. 42, 399.
- Gunasekaran, R. et al. (2009) Brain Res. 1255, 170.
- Hu, W. et al. (2009) Nat. Neurosci. 12, 996.
- Jenkins, S.M. and Bennett, V. (2001) J. Cell Biol. 155, 739.
- Jenkins, S.M. and Bennett, V. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 2303.
- Kazarinova-Noyes, K. et al. (2001) J. Neurosci. 21, 7517.
- Klein, J.P. et al. (2004) Brain Res. 1000, 102.
- Krzemien, D.M. et al. (2000) J. Comp. Neurol. 420, 70.
- Lee, A. and Goldin, A.L. (2009) Channels (Austin) 3, 171.
- Lorincz, A. and Nusser, Z. (2008) J. Neurosci. 28, 14329.
- Ohira, K. et al. (2010) Nat. Neurosci. 13, 173.
- Osorio, N. et al. (2005) J. Physiol. 569, 801.
- Royeck, M. et al. (2008) J. Neurophysiol. 100, 2361.
- Sasaki, M. et al. (2006) J. Neurosci. 26, 1803.
- Southwood, C. et al. (2004) J. Neurosci. 24, 11215.
- Suzuki, A. et al. (2004) Glia 46, 274.
- von Reyn, C.R. et al. (2009) J. Neurosci. 29, 10350.
- Yao, C. et al. (2005) Life Sci. 77, 1116.