
Sepsis remains a critical healthcare issue as it can lead to multiple organ failure, with heart complications being particularly deadly. The heart struggles under sepsis due to defects in cardiac function and inflammation that damage cellular structures. The central issue? A chemical imbalance caused by calcium in the heart cells, which, when unregulated, wreaks havoc on the body.
Now, new research published in Nature Communications tells us much more about the role transient receptor potential canonical (TRPC) channel isoforms TRPC1 and TRPC6 play in this deadly scenario. By selectively ‘switching off’ TRPC1 and TRPC6, the researchers have found a way to prevent damaging inflammation and heart failure triggered by bacterial toxins. This approach, which also involves a unique interaction with a molecular partner, calmodulin (CaM), could represent a potent new avenue of treatment for septic heart failure
Heart of the Issue
In the world of critical care medicine, sepsis, or endotoxemia (ETM), remains a formidable adversary. Around 40–50% of ETM patients suffer cardiac dysfunction, also known as ETM-induced cardiomyopathy, leading to a terrifying mortality rate of up to 70–90%. One primary instigator? Lipopolysaccharide (LPS), a bacterial endotoxin that ignites inflammation and contributes significantly to ETM progression.
Cardiac dysfunction in sepsis patients is typically marked by defects in heart muscle contraction and impaired myocardial compliance, excessive cardiac inflammation, and damaged mitochondria. The disruption of intracellular calcium concentration ([Ca2+]i), a key process in heart muscle contraction, is also common. While medications that regulate Ca2+ levels could potentially mitigate this effect, there’s still a missing piece to fully understand the regulatory mechanisms controlling abnormal Ca2+ dynamics.
TRPC: What’s the Deal?
Enter TRPC channels. Unlike voltage-dependent calcium channels, TRPC proteins form a vast non-voltage-gated cation channel superfamily. They integrate various stimuli and then transmit their activity to cellular signaling pathways via Ca2+ entry or membrane depolarization. Given their pivotal role in regulating cardiac contraction and conduction under pathological conditions, they’ve attracted significant interest in sepsis research.
TRPC channels exist in two primary subgroups: TRPC1/4/5 and TRPC3/6/7. While TRPC1/4/5 channels are activated in response to intracellular Ca2+ store depletion, TRPC3/6/7 channels respond to diacylglycerol generated by specific receptor signaling. This research focuses on TRPC1 and TRPC6 in light of their potential roles in septic cardiac dysfunction.
Uncovering the Potential of TRPC
The study sheds light on the immense therapeutic potential of TRPC channels. The researchers demonstrated that Trpc1 or Trpc6 knockouts provide significant protection against LPS-induced cardiac dysfunction and prolong survival in mice. In fact, Trpc deletion markedly improved the survival of LPS-challenged mice from 0 to 70% (Trpc1) or 60% (Trpc6). How? By inhibiting Ca2+ leakage from certain cellular structures and an inflammation cascade in endotoxemic hearts.
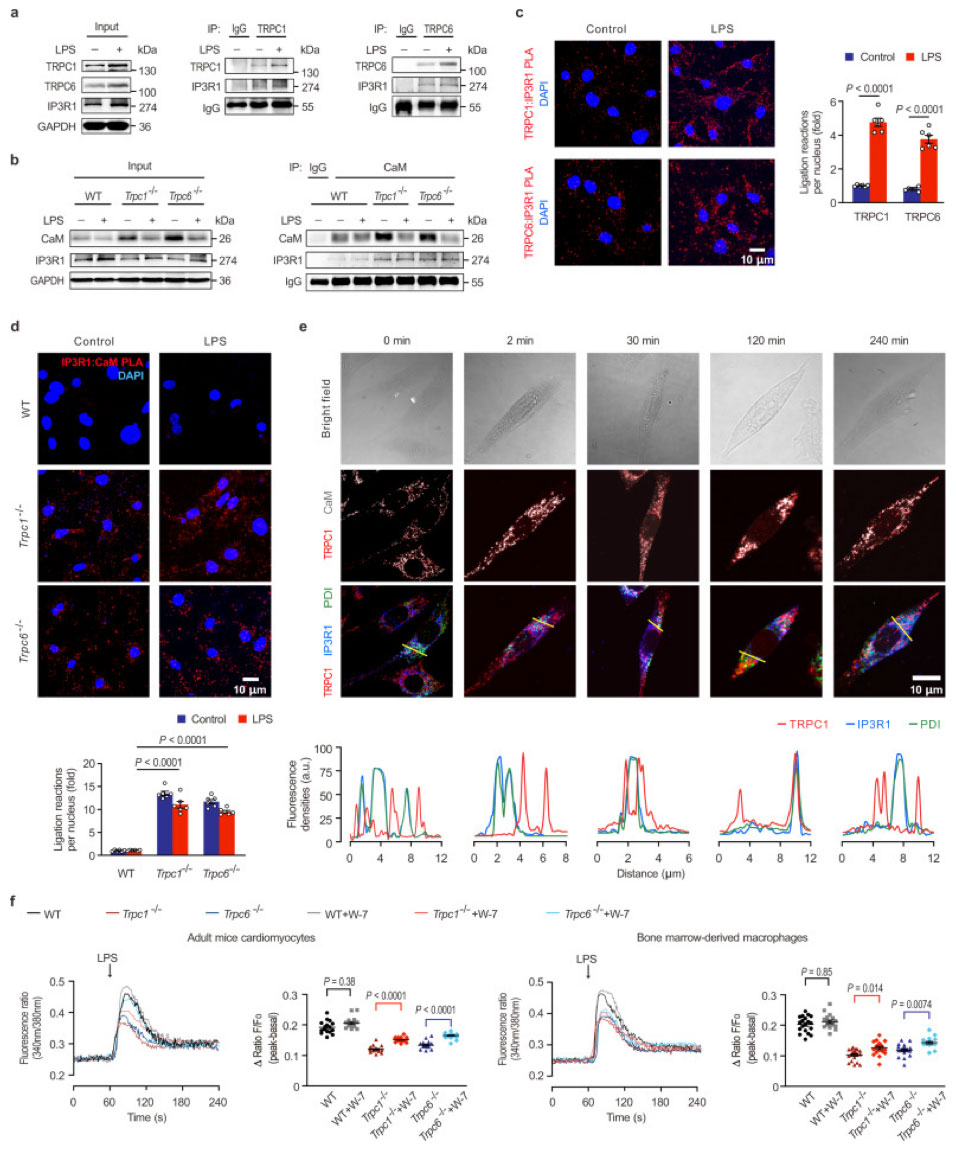
The team’s experiments revealed that TRPC1 or TRPC6 could interact with IP3R1, a receptor responsible for releasing Ca2+ from the endoplasmic reticulum (ER), a crucial cellular structure regulating intracellular Ca2+ levels. Knocking out these TRPCs significantly reduced this Ca2+ release under LPS challenge in both cardiomyocytes and macrophages. Therefore, these findings underscore the pivotal roles TRPC1 and TRPC6 play in Ca2+ mobilization in the early acute phase of septic cardiac dysfunction.
TRPC versus CaM and IP3R1: Multiple Influences
Let’s look into those TPRC-IP3R1 interactions in a little more detail. TRPC channels are capable of forming homomeric and heteromeric channels and have significant roles in signal transduction. And while TRPC1 and TRPC6 didn’t form protein complexes, they did seem to play individual roles in cardiac dysfunction related to endotoxins. So the team looked at how TRPC channels regulate CaM and calcium signaling post-LPS stimulation, specifically focusing their attention on the C-terminal of TRPC, which contains the domain that binds to CaM/IP3R.
Among all IP3R subtypes, IP3R1 was the most changed in the mouse heart. A Co-IP assay revealed that both TRPC1 and TRPC6 bind to IP3R1, without any clear interaction between CaM and IP3R1 in healthy mouse hearts. However, in the hearts of mice lacking Trpc1 or Trpc6, CaM showed a strong interaction with IP3R1, regardless of LPS treatment. This evidence suggests that LPS promotes the recruitment of IP3R1 to TRPC1 and TRPC6.
Interestingly, there was no detectable signal of CaM: IP3R1 in healthy cells, but fluorescent signals were found in the cytoplasm of Trpc1 and Trpc6 knockout cardiomyocytes (Figure 1; TRPC1 (#ACC-010, 1:100) and TRPC6 (#ACC-017, 1:100) antibodies from Alomone Labs), indicating that CaM could bind IP3R1 in the absence of Trpc. Following LPS treatment, TRPC1 co-localized with CaM increased near IP3R1, suggesting a role for both TRPC and CaM in the Ca2+ release via IP3R1.
The results reveal that IP3R1 has a higher affinity to TRPC, with CaM interacting with IP3R1 only during Trpc deficiency. When looking at the effect of the CaM antagonist, W-7, on LPS-induced Ca2+ release from ER, the team saw that W-7 had no effect on LPS-stimulated calcium levels in healthy cells, though it did significantly affect calcium levels in the absence of Trpc1 or Trpc6. The results here suggest that uncoupled CaM can inhibit LPS-stimulated Ca2+ release when TRPC is suppressed.
Implications for Treatment
Beyond their classical function as ion channels responsible for inward Ca2+ flow, this research confirms that TRPC channels integrate cytosolic calcium and inflammatory responses in septic cardiac dysfunction. TRPC deletion or blockade could prevent cardiac collapse in ETM by impeding LPS-induced intracellular Ca2+ leakage and restraining inflammatory pathways. Targeting TRPC could therefore serve as a novel therapy for cardiac dysfunction in sepsis – something potentially achievable with small molecule inhibitors.
We’ve come a long way in our understanding of TRPC’s role in ETM and this work has uncovered a new potential therapeutic target. More studies are needed to explore this exciting possibility fully, but the future looks promising. The heart of the matter? TRPC1 and TRPC6 might just be the new champions in the fight against septic cardiac dysfunction.
Antibodies
- Anti-TRPC1 Antibody (#ACC-010)
- Anti-TRPC6 Antibody (#ACC-017)
- Anti-TRPC6 (extracellular) Antibody (#ACC-120)
- Anti-TRPC2 Antibody (#ACC-027)
- Anti-TRPC3 Antibody (#ACC-016)
- Anti-TRPC3-ATTO Fluor-594 Antibody (#ACC-016-AR)
- Anti-TRPC4 Antibody (#ACC-018)
- Anti-TRPC5 Antibody (#ACC-020)
- Anti-TRPC7 (extracellular) Antibody (#ACC-066)