Over the past two decades, the ubiquitous position of Ca2+ signaling in cellular apoptosis has been carefully investigated. The body of work described here highlights the complex role played by Ca2+ in generation, regulation and coordination apoptosis.
Introduction
The word ‘apoptosis’ is derived from a Greek word meaning ‘falling leaves’ and is used to describe a form of cell death occurring in multicellular organisms. It is defined as programmed cell death that involves altruistic suicide of individual cells in favor of the organism as a whole. Apoptosis is a tightly regulated, highly efficient and energyrequiring process which engages multiple cell signaling pathways. The apoptotic network components are genetically encoded and are usually in place in a cell ready to be activated by a death-inducing stimulus1,3.
Apoptotic activity is desirable during organism development and morphological changes especially at the embryonic stage (for example the formation of digits), as well as during the activation of the immune system4. Additionally, this process is essential in the homeostasis of cell number in organs in order to determine the cell numbers of the whole organism5. Defects in apoptosis or apoptotic prevention can result in cancer, autoimmune diseases and spreading of viral infections, while neurodegenerative disorders, AIDS and ischemic diseases are caused or enhanced by excessive apoptosis5.
In contrast to the necrotic mode of cell death, apoptosis results in a loss of membrane integrity, swelling and disruption of the cell undergoing apoptosis without causing damage to the surrounding cells. Apoptotic cells can be characterized by stereotypical morphological changes which include cell shrinkage, loss of cell surface shape, chromatin condensation, DNA degradation, protein fragmentation, organelle disassembly and degradation of cells into small apoptotic bodies, which are disposed of by the immune system1.
The apoptotic process can be driven by various stimuli from outside or inside the cell; in some cases, absence of survival factors is enough to propel a cell into apoptosis, but it can also be stimulated by DNA damage, oxidative stress, treatment with cytotoxic drugs or irradiation, interruption in cell cycle signaling and death receptor ligands (TNF and Fas ligand). Cell signaling modulators, such as the universal protein kinase inhibitor Staurosporine (#S-350)6 (Fig. 1) and the tyrosine kinase inhibitor Genistein (#G-300)7 are usually used to induce apoptosis experimentally. Apoptosis occurs through two types of pathways: the death receptor pathway (extrinsic apoptotic pathways) and the mitochondrial pathway (intrinsic apoptotic pathways). The apoptotic signal involves activation of several powerful enzymes (mainly the Bcl and Caspase families). The Bcl family has a controlling role in maintaining the apoptotic signal, while the Caspase family plays a central role in cell protein degradation8,9.
Ca2+ is a fundamental second messenger in cell signaling. Stimulation of cells by Ca2+-linked signaling agents increases Ca2+ levels within the cell cytosol and the nucleus. This can modulate numerous Ca2+-regulated enzymes which have different subcellular localizations and create a wide range of spatial and temporal signals. Cytosolic Ca2+ has been implicated in the activity of Ca2+/calmodulin-dependent protein kinase (CaMK), protein kinase A (PKA) and C (PKC), mitogen-activated protein kinase (MAPK), and phosphatidylinositol (PI)-3 kinase10-14. Ca2+ metabolism plays a crucial role during the propagation of apoptosis. However, the role of Ca2+ in generation of apoptosis is controversial, in view of the fact that in different systems, Ca2+ was observed as both a survival supporter and apoptosis inducer. For instance, high K+ depolarization and subsequent Ca2+ entry into the cytosol helps to sustain the survival of granule cells15,18 On the other hand, compounds that chronically elevate intracellular Ca2+, such as Ca2+ ionophores, have been observed to induce apoptosis in a variety of cell lines19-21.
Moreover, processing of the apoptotic signal, involves intracellular Ca2+ elevation as an obligatory factor for reactive oxygen generation and mitochondrial cytochrome c release, which are both considered as apoptotic triggers22.
The following paper reviews the divisive role of Ca2+ as either a positive or negative generator of the apoptotic processes and its involvement in their progression.
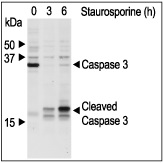
Jurkat cells were grown to 70% confluency. 2 μM Staurosporine (#S-350) or vehicle were added for 3 or 6 hours. At the end of the incubation period, the cell extracts were probed for Caspase 3 and cleaved Caspase 3 with specific antibodies.
Ca2+ Signaling: Suppressor and Promoter
Ca2+/Calmodulin-Dependent Protein Kinase in Apoptosis
Ca2+ signaling is the center of multifunctional CaMK family regulation and activation. This family, which phosphorylates a large variety of substrates, has activation properties that allow it to discriminate between Ca2+ signals that differ in spike frequency, amplitude and duration23-25. The CaMKI, II and IV subfamilies have been detected within the cell nucleus and suggested as mediators of nuclear Ca2+ signals. These kinases were implicated in control of gene transcription since they phosphorylate several transcription factors26. Several reports have indicated that both kinases negatively modulate apoptosis27,28. For instance, CaMKII up-regulation increased the resistance of glioma cells to apoptosis and KN-93, a CaMKII inhibitor, annulled this effect. In cerebellar granule cells, Ca2+ entry through L-type voltage gated Ca2+ channels supported neuronal survival in a CaMKIV-dependent manner29,30. During apoptosis, CaMKIV was reported to be negatively controlled by specific cleavage caused by caspase-3-like activity30. Moreover the serine-threonine protein kinase B, which is positively activated by the CaMKs-dependent protein kinase pathway, plays an important role in neuronal survival9. Similarly, in neuronal NG108 cells, expression of a peptide located in the noncatalytic N-terminal domain of CaMK-like kinase facilitated apoptosis (Figure 2)31,32.
Nuclear Transcription Factor-κB in Apoptosis
Intracellular Ca2+ is a coordinating factor that positively regulates the activity of nuclear transcription factor-κB (NF-κB). This transcription factor is considered an anti apoptotic agent and plays a key role in cell survival by up-regulating expression of several apoptosis inhibitor genes and negative regulating the activity of caspase-333-35. Several reports illuminate the function of this intracellular pathway in prevention of apoptosis, for instance: In hippocampal pyramidal neurons, activation of glutamate receptors leads to membrane depolarization and Ca2+ influx followed by activation of NF-κB36. In pancreatic β- cells, depolarization and L-type Ca2+ influx induce MEK/ERK dependent NF-κB related survival37.
Protein Phosphatase – Calcineurin in Apoptosis
Cytosolic Ca2+ increase has a pivotal role in activation of the serine threonine Ca2+ -calmodulin-regulated phosphatase – calcineurin (also called protein phosphatase 2B). This phosphatase is a critical transducer of Ca2+ signals in most cell types particularly in the immune system and in the heart, due to its specific responsiveness to sustained low frequency Ca2+ signals. Calcineurin was suggested to be both a promoter and a supportive agent during apoptosis.
Sustained increase in cytosolic Ca2+ has been reported to induce apoptosis in some susceptible cell types through activation of calcineurin38-40. High level forced activity of this phosphatase induced cytochrome c/caspase-3-dependent apoptosis in neurons4 and treatment with calcineurin inhibitors Cyclosporin A (#C-900) and FK-506 (#F-900) reduced susceptibility to apoptosis41. Additionally, the apoptosis-associated tyrosine kinase (AATYK) is highly phosphorylated during the normal cell cycle. Activation of calcineurin in response to high extracellular K+ mediated L-type Ca2+ influx, dephosphorylates AATYK and leads to apoptotic cell death42.
In hippocampal neurons, L-glutamate induced Ca2+ influx activates calcineurin which triggered transcription-independent apoptosis by mitochondrial targeting of BAD- a pro-apoptotic protein member of the Bcl-2 family. The Ca2+-induced dephosphorylation of BAD was correlated with its dissociation from the cytosol, translocation to mitochondria and enhancement of BAD Bcl-xL heterodimerization promoting apoptosis39.
Some reports suggest that the Ca2+-calcineurin pathway is critical in the progression of heart failure by regulating cardiomyocyte apoptosis. Moreover, induced ischemia caused more apoptosis in the hearts of transgenic mice expressing high levels of calcineurin than in the hearts of wild-type mice. Pharmacologic inhibitors of calcineurin activity block hypertrophy in vivo and in vitro and some of them (Cyclosporine A, FK-506 and Rapamycin (#R-900)) (Figure 3) were even suggested as candidates for treatment of heart disease43-44.
On the other hand, Ca2+-calcineurin activation by 2-deoxyglucose and Staurosporine prevents apoptosis of cardiac myocytes45. Calcineurin, which dephosphorylates the transcription factor NF-AT3, enables it to translocate into the nucleus, leading to prevention of apoptosis both in vitro and in vivo. Furthermore, calcineurin and protein kinase C suppressed apoptosis induced by tumor necrosis factor, through activation of NF-κB in fibroblasts and lymphoma cells46. Finally, Ca2+-mobilizing compounds such as the Ca2+ ionophore A23187 (#A-600) or the ER Ca2+ ATPase inhibitor Thapsigargin (#T-650), which both trigger calcineurin activity, can either suppress or induce apoptosis in the same cells47. The reason for this dichotomy is unknown at present, but from the data summarized here we conclude that it depended on pathways downstream from calcineurin. This idea is supported by the fact that suppression or induction of apoptosis by opposing pathways such p38 and p44/42 mitogen-activated protein kinases are located downstream to Ca2+-activated calcineurin48.
C-Jun N-terminal Kinase in Apoptosis
A distinctive feature of apoptosis is the requirement for de novo RNA synthesis. A key transcription factor for apoptosis is c-Jun, an immediate-early gene49. Ca2+ influx has been reported to be involved in c-jun N-terminal kinase (JNK) signaling pathway mediated IL-1β-induced apoptosis. Pharmacological blockers of L- and T-type Ca2+ channels, such as dihydropyridine, suppressed IL-1β-induced c-jun phosphorylation. Treatment with Ca2+ mobilizing compounds such as A23187 and Ionomycin (#I-700) caused an amplification of IL-1β-induced JNK activation and lead to apoptosis50.
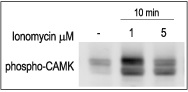
3T3-L1 cells were starved for 2h and then stimulated with 1 or 5 μM Ionomycin (#I-700) for 10 min.The cell extracts were blotted and probed with an antibody to phospho-(Thr268)-CAMKII.
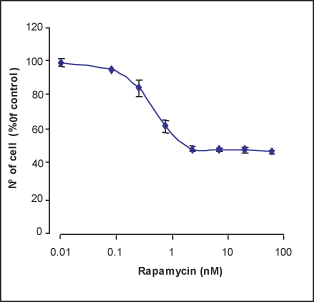
WEHI B lymphocyte cells were treated with different concentrations of Rapamycin (#R-900) for 4 days; the number of live cells was measured by XTT cell proliferation assay kit, normalized to the control (100 %) and plotted against drug concentration.
Regulation of Intracellular Ca2+ Levels and Apoptosis
Control of cytosolic Ca2+ levels involves coordination of diverse intracellular and extracellular Ca2+ currents and it plays a vital role in the initiation of the apoptotic process. Manipulation of cytosolic Ca2+ by different experimental methods may interfere with cellular process to induce apoptosis. For example, sustained increase of Ca2+ by Ca2+-mobilizing compounds such as the Ca2+ ionophore A23187 (Figure 4) and Ionomycin were reported to act as apoptosis inducers in some cell lines47,51,8.
Ligand Gated Ca2+ Permeability
Some receptors that function as ligand gated Ca2+ permeable channels play a key role in Ca2+ homeostasis and have been formally implicated in mediating apoptosis. This role is reported to be taken by the purinergic receptor P2X (P2X R)52, inositol 1,4,5-triphosphate receptor (IP3R)53. and ryanodine receptor (RyR).54 Whereas the P2XRs are cell surface membrane Ca2+ channel, the IP3R and RyR function as Ca2+ release channels in the ER membrane.
The P2XRs are non selective cation channels with significant permeability to Ca2+. They induce signaling by allowing Ca2+ entry relative to the extracellular concentration of ATP. The first clue to the involvement of P2XRs was the finding that high concentrations of ATP induced apoptosis by increasing the intracellular Ca2+level55,56. P2X1 channel proteins were up-regulated in thymocytes during dexamethasone-induced apoptotic cell death, while pharmacological inhibition of the P2XR1 protect thymocytes from apoptosis52,55. P2X channels were reported to mediate the apoptotic ATP signal by Ca2+ overload in blood, brain and skin cells57.
Intracellular organelles such as the ER and mitochondria have higher Ca2+ concentrations than the cytosol and they are considered cellular Ca2+ stores. Regulation of ER Ca2+ release controls many cellular functions. Enzymatic cascades which are dependent on Ca2+ concentration in the ER lumen integrate rapid Ca2+signaling with longlasting adaptive responses through modifications of protein synthesis and processing. Several studies have suggested a central role for participation of ER and intracellular Ca2+ levels in the regulation of mitochondrial cytochrome c release that leads to induction of apoptosis58.
IP3Rs are intracellular ion channels that mediate the release of Ca2+ from the ER and play a central role in control of intracellular Ca2+ homeostasis. Cytosolic IP3 is produced in response to GPCR stimulated phospholipase C. Binding of IP3 to the receptor induces transient opening of the receptor channel, facilitating Ca2+ flow from the ER lumen into the cytoplasm, thereby producing a transient elevation of cytosolic Ca2+ that in turn activates signal transduction kinases. Although IP3Rs are mostly located on the ER membrane, evidence suggests that they are also located on the plasma membrane, enabling the IP3R to mediate entry of extracellular Ca2+ into the cytoplasm. The IP3R has been suggested to be involved in adrenal corticosteroid related apoptosis58,59 through its downstream target, calcineurin60. Cytosolic Ca2+ levels are tightly controlled by IP3R channel activity. Intermediate Ca2+ levels facilitate the channel closure, and prevent Ca2+overload54,62.
As a response to a pathological stimulus, Ca2+ together with cytochrome c participates in self amplifying feedback to promote apoptosis61. Low levels of cytochrome c prevent the closing of the IP3R channel during Ca2+ overload, leading to a burst of Ca2+ flow from the ER lumen into the cytosol. This in turn potentiates release of all the mitochondrial cytochrome c, which is sufficient to operate the caspase cascade to promote apoptosis61.
RyRs operate as Ca2+ release channels particularly in skeletal and cardiac ER, they are also located in the cell membrane of a variety of cells and are highly expressed in T- and B-lymphocytes. These channels provide a pathway for fast Ca2+ release from intracellular stores into the cell cytosol. In some cases, RyR channels transmit Ca2+ signals directly to closely associated mitochondria54. In pancreatic β-cells RyR2 was found to be involved in buffering of Ca2+ influx during stimulation with glucose or high K+. Blocking of Ca2+ flux through the type 2 RyR in these cells markedly increased apoptosis in calpain-dependent death pathways62. On the other hand, depletion of intracellular Ca2+ stores and cytosolic Ca2+ overload via caffeine or Ryanodine (#R-500) activated RyR channels in CHO cells, observed to enhance translocation of the proapoptotic factor Bax into the mitochondrial membrane and induction of apoptosis3,63,64.
The maintenance of ER Ca2+ stores is result of equilibrium between Ca2+ release and Ca2+ refill which involves opening of cation channels as well as activity of Ca2+ pumps. The Sarco Endoplasmic Reticulum Ca2+ATPase (SERCA) is a critical molecule which pumps Ca2+ from the cytosol into the ER lumen in order to maintain low intracellular Ca2+65. Dysfunction of this pump by inhibition with Thapsigargin caused ER Ca2+depletion and cytosolic Ca2+ overload, followed by extensive DNA fragmentation and apoptosis in a variety of cells (Figure 5)66. Increase of the cholesterol phospholipid ratio which occurs under pathological conditions changes the fluidity of the ER membrane and inhibits SERCA. This effect can be avoided by pharmacologic manipulations that block cholesterol trafficking to the ER67.
Ca2+ Channels
Ca2+ influx in nonexcitable cells regulates such diverse processes as exocytosis, enzyme activity, gene regulation and cell proliferation. Overload of extracelluar Ca2+ can damage tissues, this effect may attribute to apoptotic effect caused by Ca2+ influx through L-type voltage-dependent Ca2+ channels and by Transient Receptor Potential (TRP) non selective ion channels.
L-type Ca2+ channels were exclusively upregulated as a response to chronic cell membrane high K+ depolarization, leading to cell apoptosis in some cell lines68. Such an observation might be explained by the fact that a steep rise in intracellular Ca2+ is buffered to some degree by mitochondrial Ca2+ uptake. However, a continuous increase in cytosolic Ca2+, such as in the case of channel upregulation, revokes the buffering capacity and causes dysfunction of these organelles69-71. On the other hand, high K+ induced transmembrane Ca2+– influx was observed to be essential in the maintaining of nerve and renal cells since blockage of Ca2+influx might prevent cell survival. Moreover, the L-type Ca2+ channel agonist – (±)-Bay K8644 (#B-350) is a potent agent that prevents nerve cell loss during low K+ enhanced apoptosis (Figs. 6-7)72-74.
The TRPM2 channels which belong to the TRP family were shown to contribute to apoptotic cell death. This Ca2+ permeable channel was reported to be gated by adenosine diphosphate-ribose (ADP-ribose) and H2O2 which are both evoked during oxidative stress, resulting in a disruption of intracellular free Ca2+homeostasis75,76.

Jurkat cells were grown to 70% confluency. A23187 (#A-600) (2 μM) or vehicle were added to cell culture for 6 hours. At the end of the incubation, the cells were extracted and blotted. Apoptosis was probed by detection of cleaved Caspase 3 using Caspase 3 specific antibodies.
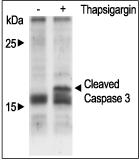
Jurkat cells were grown to 70% confluency. Then 1 μM Thapsigargin (#T-650) or vehicle were added for 6 hours. At the end of the incubation period, the cells extracts were detected for cleaved Caspase 3 with Caspase 3 specific antibodies.
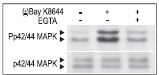
Cells were preincubated with or without 2 mM EGTA to chelate Ca2+ for 20 min then stimulated with 50 μM (±)-Bay K 8644 (#B-350) for 10 min. The cell proteins were resolved by SDS PAGE and probed with Anti-Phospho-p42/44 MAPK (upper panel) or with Anti-p42/44 MAPK (lower panel).
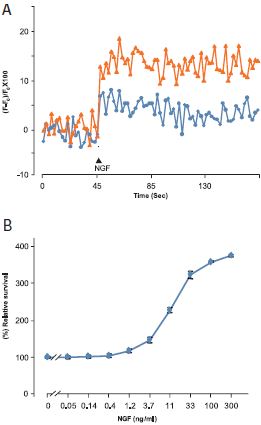
A. Ca2+ traces from Fura-2 AM loaded PC12 cells treated with 300 ng/ml Native mouse NGF 2.5S protein (99%) (#N-240) in presence (blue line) or absence (orange line) of 300 nM L-Type Ca2+ channel blocker- Calcicludine (#C-650, replaced with #SPC-650). B. PC12 cells were growth in absence of serum (to induce apoptosis) and in presence of different concentrations of Native mouse NGF 2.5S protein (99%). The cell survival as function of NGF concentration was measured after 4 days using the XTT -method calculated as relative percentage of the control without NGF and plotted against NGF concentrations.
Extracellular Ca2+ Sensing Prevents Apoptosis
Ca2+ sensing plays a key role in regulation of the calcitrophic hormone PTH in tissues that maintain systemic Ca2+ homeostasis. The G protein-coupled extracellular Ca2+-sensing receptor (CaR) has been elucidated as responsible for recognizing and responding to small changes in extracellular Ca2+ level. It is becoming clear that the CaR also participates in a variety of non-Ca2+ homeostatic functions, such as control of ion channels, hormone secretion, and regulation of cell-cycle events77,78. The role of CaR in apoptosis has been demonstrated when it was shown that a rise in extracelluar Ca2+ protected AT-3 prostate carcinoma cells and CaR-transfected HEK293 cells from apoptotic cell death. Furthermore, anti apoptotic effect of extracellular Ca2+was also observed in c-myc overexpressing/serum deprived cells (all the cells were shown to express CaR)79. PI3K and p38 MAPK were suggested to be the downstream pathways that mediate the mitogenic response to extracellular Ca2+ activated CaR80. However, the downstream mechanism by which CaR prevents apoptosis has not been proved yet.
Ca2+ and Mitochondrial Apoptosis
Ca2+ plays a central role in mitochondrial function and mitochondrial regulation and it acts on several levels within the organelle to stimulate ATP synthesis. Although the luminal level of Ca2+ in mitochondria is high, most of the mitochondrial effects of Ca2+ require its entry across the double membrane into the matrix. The mitochondrial outer membrane was thought to be permeable to Ca2+, even though recent evidence suggests that the outer membrane Voltage-Dependent Anion Channel (VDAC) is a ruthenium red-sensitive Ca2+channel. This Mitochondrial Permeability Transition (MPT) pore was also proposed to conduct non specific low molecular insults movement81,82. However, dysregulation of mitochondrial Ca2+ homeostasis is now recognized to play an active role in apoptosis by switching the mitochondrial Ca2+ signaling to facilitate Ca2+-induced opening of the MPT pore81-83. For example, mitochondrial matrix Ca2+ overload enhances generation of Reactive Oxygen Species (ROS) which trigger the opening of the MPT pore and release of apoptogenic factors such as cytochrome c59. Because mitochondrial cytochrome c release during apoptosis is an “all-or-nothing” event occurring within a rapid time frame3, it has been suggested that the Ca2+ spike coordinates cytochrome c release by regulation of the VDAC opening84. This paradigm is supported by the observation that increased expression of this channel augments the mitochondrial sensitivity to ER-stress caused by thapsigargin-induced apoptosis85-87,22.
The primary ROS manufactured by the mitochondria is superoxide (O2–), which is converted to H2O2 either by spontaneous dismutation or by the enzyme superoxide dismutase22. Apoptosis is reported to be accompanied by a burst of ROS which damages cellular components such as proteins, lipids, and DNA88. At the heart of understanding how Ca2+ can be both a physiological and a pathological effector of mitochondrial function is the fact that Ca2+ modulates mitochondrial ROS generation. The normal production of ROS is limited only to the amount required for microdomain cell signaling89 whereas overload of mitochondrial Ca2+ during pathological conditions upregulates the production of QH – a ROS precursor, thereby causing an increase in ROS production. Alternatively, Ca2+ can stimulate nitric oxide generation5, another generator of ROS, to generate apoptosis90.
Conclusion
Apoptotic processes are an integral part of the life cycle: development, differentiation, proliferation, homoeostasis, regulation and function of the body systems and in the removal of defective and therefore harmful cells. Thus, dysfunction or dysregulation of the apoptotic program is implicated in a variety of pathological conditions. Due to its importance in such varied biological processes, programmed cell death is a widespread phenomenon investigated in many areas of biology.
Pathological elevation in cytosolic Ca2+ concentrations (above 10 μM) is deleterious and can modulate apoptotic cell death through multiple mechanisms in a specific cells. Understanding the dual role played by Ca2+ signaling in initiation of apoptosis is relevant in many clinical situations such inherited muscle diseases and autoimmune diseases91,5.
In view of data summarized here we conclude that different Ca2+ stimuli have distinct effects on stimulation of apoptotic process, dependent on cell type, cell environment, Ca2+ source and Ca2+ levels. Furthermore, cell apoptosis is strongly dependent on the biochemical situation or pathways that are already activated at the time of the apoptotic stimulus. Moreover, we speculate that Ca2+ can serve as either positive or negative trigger, but not as an exclusive factor, for driving apoptosis. The final result will be dictated by the cell situation, which is an integration of the levels, availability and location of certain cell components as well as equilibrium among different signaling pathways that take place during apoptosis modulation.
On the other hand, during progression of the apoptosis process, Ca2+ is an active factor that promotes the conversion of proapoptotic components into apoptotic conformation.
References
- Ellis, R.E. et al. (1991) Annu. Rev. Cell Biol. 7, 663.
- Ishizaki, Y. et al. (1995) Mol. Biol. Cell. 6, 1443.
- Weil, M. et al. (1996) J. Cell Biol. 133, 1053.
- Rathmell, J.C. et al. (1998) J. Exp. Med. 188, 651.
- Rathmell, J.C. and Thompson, C.B. (2002) Cell 109, 97.
- Martin, S.J. and Green, D.R. (1995) Cell. 82, 349.
- Linford, N.J. et al. (2001) J. Pharmacol. Exp. Ther. 299, 67.
- Widmann, C. et al. (1998) J. Biol. Chem. 273, 7141.
- Alnemri, E.S. et al. (1996) Cell 87, 171.
- Frodin, M. et al. (1995) J. Biol. Chem. 270, 7882.
- Finkbeiner, S. and Greenberg, M.E. (1998) J. Neurobiol. 37, 171.
- Persaud, S.J. et al. (1996) Biochem. J. 313, 119.
- MacFarlane, W.M. et al. (1997) J. Biol. Chem. 272, 20936.
- Rhodes, C.J. (2000) J. Mol. Endocrinol. 24, 303.
- Gallo, V. et al. (1987) J. Neurosci. 7, 2203.
- Boutillier, A.L. et al. (1999) Eur. J. Neurosci. 11, 441.
- Mattson, M.P. and Chan, S.L. (2003) Nat. Cell Biol. 12, 1041.
- Wonderlin, W.F. (2002) Biophys. J. 82, 645.
- Jiang, S. et al. (1994) Exp. Cell Res. 212, 84.
- Martikainen, P. et al. (1991) Cancer Res. 51, 4693.
- Reynolds, J.E. and Eastman, A. (1996) J. Biol. Chem. 271, 27739.
- Foyouzi-Youssefi, R. et al. (2000) Proc. Natl. Acad. Sci. USA 97, 5723.
- Sheng, M. et al. (1990) Neuron 4, 571.
- Finkbeiner, S. and Greenberg, M.E. (1996) Neuron 16, 233.
- Corcoran, E.E. and Means, A.R. (2001) J. Biol. Chem. 276, 2975.
- Heist, E.K. and Schulman, H. (1998) Cell Calcium 23, 103.
- Anderson, K.A. et al. (1997). Mol. Endocrinol. 11, 725.
- Hajimohammadreza, I. et al. (1995) J. Neurosci. 15, 4093.
- Yang, B.F. et al. (2003) J. Biol. Chem. 278, 7043.
- Yano, S. et al. (1998) Nature 396, 584.
- McGinnis, K.M. et al. (1998) J. Biol. Chem. 273, 19993.
- Kruidering, M. et al. (2001) J. Biol. Chem. 276, 38417.
- Miyamoto, S.S. et al. (1998) Mol. Cell Biol. 18,19.
- Kanno, T. and Siebenlist, U. (1996) J. Immunol. 157, 5277.
- Sun, L. and Carpenter, G. (1998) Oncogene 16, 2095.
- Mattson, M.P. et al. (2000) J. Neurochem. 74, 443.
- Bernal-Mizrachi, E. et al. (2002) Diabetes. 51, S484.
- Wang, H.G. et al. (1999) Science 284, 339.
- Shibasaki, F. and McKeon, F. (1995) J. Cell Biol. 131, 735.
- Asai, A. et al. (1999) J. Biol. Chem. 274, 34450.
- Zacharchuk, C.M. et al. (1990) J. Immunol. 145, 4037.
- Tomomura, M. and Furuichi, T. (2005) J. Biol. Chem. 280, 35157.
- Ankarcrona, M. et al. (1996) FEBS Lett. 394, 321.
- Saito, S. et al. (2000) J. Biol. Chem. 275, 34528.
- Molkentin, J.D. et al. (1998) Cell 93, 215.
- Van Antwerp, D. J. et al. (1996) Science 274, 787.
- Lotem, J. et al. (1999) Proc. Natl. Acad. Sci. USA 96, 12016.
- Chiang, L. W. et al. (2001) Proc. Natl. Acad. Sci. USA 98, 2814.
- Størling, J. et al. (2005) Endocrinology. 146, 3026.
- Squier, M.K. et al. (1994) J. Cell Physiol. 159, 229.
- Chvatchko, Y. et al. (1996) Immunity 5, 275.
- Khan, A.A. et al. (1996) Science 273, 503.
- Pizzo, P. et al. (1991) Biochem. J. 274, 139.
- Boehning, D. et al. (2003) Nat. Cell Biol. 5, 1051.
- Johnson, J.D. et al. (2004) FASEB J. 18, 878.
- Zheng, L.M. et al. (1991) J. Cell Biol. 112, 279.
- Greig, A.V.H. et al. (2003) J. Invest. Dermatol. 121, 1145.
- Berridge, M.J. (1993) Nature 361, 315.
- Distelhorst, C.W. and Dubyak, G. (1998) Blood 91, 731.
- Jayaraman, T. and Marks, A. R. (2000) J. Biol. Chem. 275, 6417.
- Marks, A.R. (1997) Am. J. Physiol. Heart Circ. Physiol. 272, 597.
- Johnson, J.D. et al. (2004) J. Biol. Chem. 279, 24794.
- Pan, Z. et al. (2001) J. Biol. Chem. 276, 32257.
- Pan, Z. et al. (2000) J. Biol. Chem. 275, 19978.
- East, J.M. (2000) Mol. Membr. Biol. 17, 189.
- Jiang, S. et al. (1994) Exp. Cell Res. 212, 84.
- Shigehito, N. et al. (2003) J. Biochem. (Tokyo), 134, 359.
- Cano-Abad, M.F. et al. (2001) J. Biol. Chem. 276, 39695.
- Lieberthal, W. and Nigam, S.K. (1998) Am. J. Physiol. 275, F623.
- Rana, A. et al. (2001) Apoptosis 6, 83.
- Lieberthal W. et al. (1998) Semin. Nephrol. 18, 505.
- Gallo, V. et al. (1987) J. Neurosci. 7, 2203.
- Collins, F. and Lile, J.D. (1989) Brain Res. 502, 99.
- Tanaka, T. et al. (2004) J. Am. Soc. Nephrol. 15, 2320.
- Wehage, E. et al. (2002) J.Biol. Chem. 277, 23150.
- Hara, Y. et al. (2002) Mol. Cell 9, 163.
- Brown, E.M. et al. (1993) Nature 366, 575.
- Brown, E.M. and MacLeod, R. J. (2001) Physiol. Rev. 81, 239.
- Lin, K.I. et al. (1998) Biochem. Biophys. Res. Commun. 249, 325.
- Tfelt-Hansen, J. et al. (2004) Endocrinology 145, 1211.
- Gincel, D. et al. (2001) Biochem. J. 358, 147.
- Hodge, T. and Colombini, M. (1997) J. Membr. Biol. 157, 271.
- Halestrap, A.P. et al. (2002) Biochimie 84, 153.
- Brookes, P.S. et al. (2004) Am. J. Physiol. Cell Physiol. 287, C817.
- Martinou, J.C. et al. (2000) Nat. Cell Biol. 2, E41.
- Boehning, D. et al. (2004) Cell Cycle 3, 252.
- Crompton, M. (2000) J. Physiol. (Lond.) 529, 11.
- Halliwell, B. and Gutteridge, J.M.C. (1999) Oxford: Oxford Univ. Press, 3rd ed.
- Castilho, R.F. (1995) Free Radic. Biol. Med. 18, 479.
- Brookes, P. and Darley-Usmar, V. M. (2002) Free. Radic. Biol. Med. 32, 370.
- MacLennan, D.H. (2000) Eur. J. Biochem. 267, 5291.