Involvement of ion channels in apoptosis is linked to critical aspects of this complex cellular process such as coordination of the Ca2+ signal, cell shrinkage and mitochondrial integrity. The body of work described below demonstrates the large variety and critical involvement of ion channels in this cellular process. It highlights the emerging roles discovered for this diverse group of membrane proteins in non excitable tissue physiology and pathophysiology.
Introduction
All cells of multi-cell organisms contain a genetic program ready to be activated for their death, with the actual execution of this program called apoptosis. Apoptotic pathways include several proteins with powerful enzymatic cell degrading potential (mainly belonging to the Bcl-2 and caspase families, and cytochrome C). These proteins are latent most of the cells’ life (either by compartmentalization or by co factor inhibition). The latent proteins are activated once a “death” signal has been consolidated by the cells’ existing signaling mechanisms. The program also includes elements of environmental awareness as the degraded cells remaining are packed and disposed of (for review see reference 1).
Apoptosis is a very important factor in an organism’s development, especially at the embryonic stage and in normal renewing tissue homeostasis (for example in epithelial and red or white blood cells)2. On the other hand the same mechanism is activated in pathophysiological conditions such as neuronal or cardiac ischemia (see for example3). In addition, apoptosis prevention might be one of the necessary steps for the transformation of normal to cancerous cells4.
Time course and stages of apoptosis progression includes a number of well-defined cellular and morphological changes5. These include cell shrinkage or apoptotic volume decrease (AVD), nuclear condensation, DNA fragmentation and formation of sub-cellular apoptotic bodies, that undergo phagocytosis by neighboring cells. Processing of the apoptotic stimulus, involve Ca2+ signals that lead to disruption of mitochondrial membranes. The latter leads to the release of proapoptotic factors, activating the caspase machinery that degrades the cell6. In parallel to stimulus processing, the cell shrinks, involving the participation of ion channels in the secretion of salts5.
Methods of experimental induction of apoptosis usually mimic certain cellular process that serve as an apoptosis induction mechanism. Nevertheless, many factors bypass certain cellular pathways that are upstream of the point where they interfere with cellular function. For example, the build-up of cytoplasmic Ca2+concentration probably plays a key role in the convergence of the initial apoptotic signal6. Many apoptotic insults involve different ways of chronic cytoplasmic Ca2+ elevation. Some include inhibition of cytoplasmic Ca2+ sequestering into ER, like Thapsigargin7, while others may perforate the membrane specifically for Ca2+by the use of a ionophore, like A231878. Apoptotic inducers may be as diverse as global kinase inhibition (Staurosporine)9 or Hydrogen Peroxide (H2O2), mimicking the reactive oxygen species (ROS)3 produced during apoptosis. However, in many cell types apoptosis is signaled by external ligands of death receptors (such as TNF-α or Fas ligands), which in many cases were used to induce experimental apoptosis (see table)10.
Ion channels are integrated membrane proteins which are exposed on both sides of the membrane. These proteins allow specific ions to cross membranes down their electrochemical gradient, usually in response to an appropriate stimulus. Ion channels are known to control a number of cellular processes including:
- Change in a given ion concentration.
- Changes in membrane potential (due to their charge being translocated across the membrane).
- Changes in the cell osmotic balance.
- Mediators of a cell’s response to a wide range of both intra- and extra-cellular chemical and/or physical stimuli11.
Ion channels that have been implicated in apoptosis as regulators of local and global ion content belong to a wide range of channel families. The channels may differ regarding the ions toward which they are permeable, the stimulus that opens the channel, localization of the channel (i.e. plasma membrane, mitochondria etc.) and their overall role as pro- or antiapoptotic agent (see table).
These channels were reported to contribute to one of the following:
- Cytosolic Ca2+ increase, leading to activation of Ca2+ dependent apoptotic machinery.
- Ion fluxes between mitochondria and cytosol, resulting in either initiation of apoptosis or protection of apoptosis stimulated cells.
- Permeation to large molecules such cytochrome C, facilitating their translocation from mitochondria to cytosol.
- K+ and Cl– efflux (from the cytosol to the extracellular space), leading to and accompanying water efflux and cell shrinkage, also leading to reduction in cytosolic K+ and relief of apoptotic inhibition.
Below, we will describe the possible roles played by specific ion channels in mediating processes crucial for the apoptotic signal propagation, either as mediators of the insult or as effectors activated by upstream stimuli.
Ca2+ permeable channels on plasma membrane
Ca2+ homeostasis plays a crucial role in apoptosis. It involves the generation, amplification and coordination of a diversity of Ca2+ signals by means of interplay between Ca2+ stores6. One such Ca2+ store is the extracellular fluid, which could be accessed by the opening of Ca2+ permeable channels located on the plasma membrane. This role in apoptosis is reported to be taken by either P2X (ionotrophic ATP receptor activated by binding of extracellular ATP resulting in the opening of a non selective, Ca2+ permeable cation channel), TRP (Transient Receptor Potential, non-selective cation channels, activated by different stimuli) or by CaV (voltage dependent Ca2+ selective) channels.
P2X channels are the direct receptor of the apoptotic insult (ATP12) as well as the transducer of the ATP signal into elevation in Ca2+ in the cytoplasm. Therefore, these channels demonstrate the role played by Ca2+ influx as one of the first events which eventually lead to cell death. P2X7 channel was reported to mediate ATP induced apoptosis in blood and bone marrow derived13-17, as well as in skin cells18. The specific contribution of the P2X7 channel was assessed both with specific pharmacology and by the use of a natural dominant negative mutant. In thymocytes, ATP induced apoptosis, which resulted in elevated P2X1 channels19. In neurons, P2X4 mRNA levels were elevated together with P2X7 following a starvation insult20. This demonstrates the difference between cells that readily express this ATP receptor channel (blood cells), making them vulnerable to external ATP, compared to cells that express the channel only in response to stress (neurons).
L-type voltage dependent Ca2+ (CaV1 subfamily) channel currents in chromaffin cells, were instantly upregulated as a response to high extracellular K+ insult (depolarization of the cell membrane among other effects), leading to apoptotic cell death21. Such an observation places these voltage activated channels as first order effectors transforming membrane depolarization into Ca2+ influx that results in apoptosis. However, a longer process involving elevated channel expression levels was suggested in physiological context. CaV1 channels have been implicated as apoptosis mediators in insulin producing cells in response to diabetic patient’s serum22. In pancreatic β cells CaV3 (T-type) channels were upregulated in response to cytokines (that are probably contained within diabetic patients’ serum)23.
TRP channels were suggested to be involved in apoptotic Ca2+ signaling and as direct receptors of certain insults. Two channels belonging to the TRPM subfamily of TRP cation channels were shown to mediate apoptotic insults, by conducting Ca2+ into cells: TRPM2 in tumor cell lines24,25 and TRPM7 in neurons that were protected pharmacologically from other forms of cell death3. The latter was motivated by failure to protect against neuronal ischemic insults by blocking the excitotoxicity mediators CaV and ionotrophic glutamate receptors. The TRPM Ca2+ currents develop slowly in response to apoptosis induction. However, the slow time course might represent the build-up of ROS in response to the oxygen-glucose deprivation insult, since TRPM channels are gated directly by H2O23.
In experiments where apoptosis was stimulated by particulate matter (PM), mimicking air pollution, the vanilloid receptor (TRPV1 channel) was found to mediate apoptosis in epithelial airways cells and sensory neurons26. External expression of the Drosophila TRPL channel (homologue of the TRPC channel subfamily), supported apoptosis in prostate cancer cell line27.
The initial build-up of cytosolic Ca2+ described above, may serve as a messenger for many cellular agents and process. For the purpose of this review we should consider Ca2+ activated channels, such as KCa and ClCa channels on the plasma and/or mitochondria and the IP3 receptor (IP3R) on the ER membrane.
Intracellular channels on ER and mitochondrial membrane
Ca2+ signal convergence results in activation of intracellular channels that leads to cytochrome C release from mitochondria6. This section will focus on a few more Ca2+ channels as well as on channels contributing to mitochondrial integrity.
IP3R are Ca2+ activated Ca2+ channels, on the ER membrane, which facilitate Ca2+ flow from ER to the cytoplasm, once the cytoplasmic Ca2+ level is intermediate (i.e. a bell shaped dependency in which the channel is closed both when Ca2+ is in the resting level and during Ca2+ overload) and plays a key role in Ca2+ homeostasis. It is also recruited to participate in an inter-organellar Ca2+ cytochrome C self amplifying signal, which is critical for downstream apoptotic mechanisms6,28. Cytochrome C can enhance IP3R activity as it binds the IP3R with very high affinity and prevents channel closure during Ca2+ overload28. During apoptosis, small amounts of mitochondrial cytochrome C translocate to the ER (a process that depends on IP3R activity) and potentiate IP3R. This leads to a bursting Ca2+ overload, which coordinates cytochrome C release from all the cell’s mitochondria, leading to activation of the caspase cascade28.
The events linking the ER released Ca2+ to cytochrome C release happen in the mitochondria and involve channels on both the outer and inner mitochondrial membranes (OMM and IMM respectively). The sequence of events and the exact roles played by particular channels are not fully resolved. A Ca2+ channel sensitive to Ruthenium Red blockade was recently suggested as the Ca2+ route into mitochondria. However, the exact role of the channel in apoptosis as well as its molecular identification is not yet clear30. In addition, other channels are involved in the maintenance/disruption of IMM potential and the release of cytochrome C from its “resting” location (between OMM and IMM) into the cytosol.
Bcl-2 proteins, an established apoptotic protein family, are suggested to act both as modulators of other channels and as channels themselves29, 31. The latter activity carried out by the Bcl-2 protein BAX, was suggested to generate the transducer of cytoplasmic Ca2+ elevation into cation currents disrupting the IMM potential. Other Bcl-2 proteins have been suggested to form the channel that conducts cytochrome C out of the mitochondria (for further details see32).
However, it is more accepted that VDAC1 channels on the OMM (which are modulated by Bcl-2 proteins) actually mediate cytochrome C translocation33. VDAC channels are big pores, with complex behavior that are usually considered as voltage dependent anion channels. VDAC channels, however, may play a protective role, as was shown for the VDAC2 isoform that is an inhibitor of Bak oligomerization and therefore it helps keep this proapoptotic protein in chains34. IMM depolarization is an important factor leading to VDAC1 priming to serve as cytochrome C translocator. IMM depolarization is caused by dissipation of H+ gradient, probably with the participation of the Cl- intracellular channel, CLIC435, 36.
The dissipation of H+ gradient could be compensated for by activation of K+ selective channels on the IMM. These include an ATP sensitive (KATP, which is a channel formed by a yet unidentified protein)37, 38 and a Ca2+ sensitive (KCa1.1)39 K+ channels. Mitochondrial matrix ATP levels drop and Ca2+ load activates the two channels, respectively, with these two events accompanying H+ gradient dissipation. This protective pathway (involving a not fully resolved mechanism) is suggested to contribute to protective physiological phenomenon and is exploited by specific pharmacological augmentation of KATP to protect hearts during surgery38.
Thus, ion channels localized on intracellular organelle membranes play a key role in consolidation of the initial apoptotic signal. However, other channels may serve to dissolve such signals and protect cells challenged by an insult.

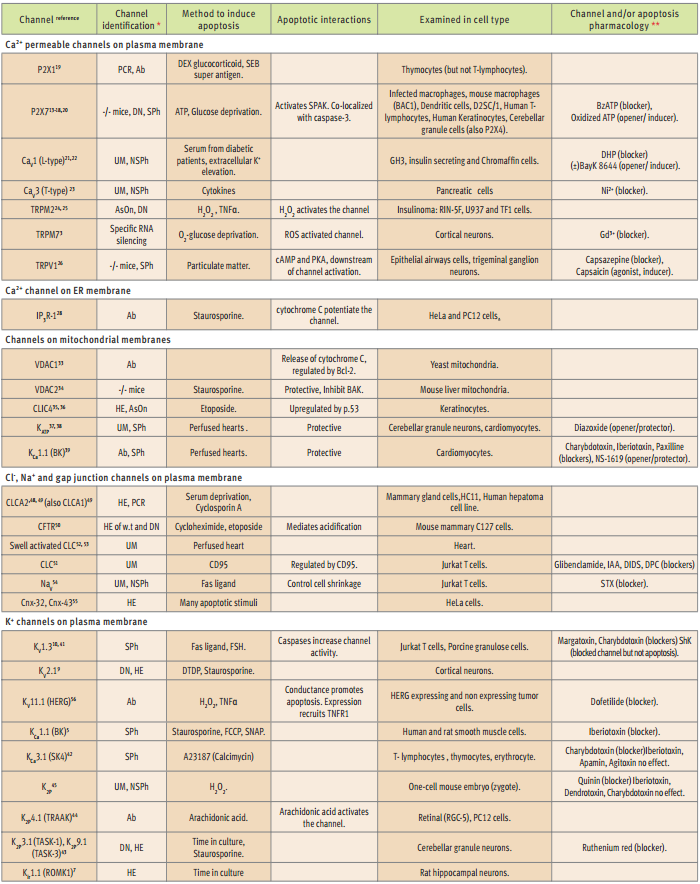
*DN= dominant negative mutant, UM = unidentified molecularly, HE = heterologous expression, Ab = specific antibody, SPh = specific pharmacology, NSPh = non specific pharmacology, AsOn = antisense oligonucleotide, PCR = Polymerase Chain Reaction, w.t. = wild type, **blocker = of both channel and apoptosis, opener/inducer = of channel/apoptosis.
Plasma membrane K+ efflux channels
K+ homeostasis, (high cytoplasmic compared to the low external concentration) is disrupted during apoptosis. That is, K+ ions must leave the cell as an obligatory step in the apoptotic pathway. However, the exact mechanism by which K+ depletion promotes apoptosis is not fully resolved. The activity of some apoptotic enzymes was suggested to depend on low K+, perhaps explaining the necessity of K+ depletion in apoptosis. K+ depletion is strongly linked to cell shrinkage, a hallmark of, but also a possible active contributor to apoptosis5. Therefore, upregulation of the activity of K+ channels probably lie in the basis of increased K+outflow, critically contributing to apoptosis5, 40.
Several reported observations suggest that any K+ channel is good for the job of emptying the cell of its K+ ions. This implies that following apoptotic stimulation the cell recruits any available K+ channels, in the context of the apoptotic stimulus and possible changes in expression profiles. Three arguments support this idea:
1. K+ channels belonging to four structurally distinct channel families were reported to be involved in apoptosis (see table).
2. There is correlation between specific channel blockers and prevention of apoptosis in different cell types41,42.
3. Heterologous expression of several types of K+channels primed the same cell type (for example, cultured hippocampal neurons7,43) to apoptosis.
It is hard to correlate the specific K+ channel that is activated with a specific apoptotic insult. For example, Staurosporine induced apoptosis that was mediated by KV2.1 channels in neurons9, but in smooth muscle cells it was mediated by KCa1.1 channels. The latter mediated also other apoptotic stimuli in these cells5.
Phenotypic differences between cell types might be correlated to the specific K+ channel they are using in apoptosis. For example, while expression of the two pore domain channel K2P9.1 (TASK-3), is essential for apoptosis in cerebellar granule neurons43, the voltage dependent KV2.1 carries out the same task in cortical neurons9. Most granule neurons are eliminated with cerebellum maturation shortly after birth and die in culture, while cortical neurons can be maintained for long periods in primary cultures. This difference may be attributed to the particular K+ channel that the cell uses to mediate apoptosis. It is interesting to note that overexpression of K2P9.1 in hippocampal neurons induced apoptosis and arachidonic acid activated K2P4.1 (TRAAK) to induce apoptosis in PC12 cells43, 44.
Since K+ channels are crucial to apoptosis, specific channel blockers are very indicative tools, especially as these also blocked the death process (see table)41, 42. For example, Iberiotoxin, a specific blocker of KCa1.1 channels blocks apoptosis in smooth muscle cells5, but failed to influence such a process in T-lymphocytes42and in a mouse embryo cell45. In these two cell types apoptosis was carried out by another Ca2+ dependent channel, KCa3.1 or by a K2P channel, respectively. Charybdotoxin, a less specific K+ channel blocker, was effective in blocking apoptosis in granulose cells as did Margatoxin, pointing to the involvement of KV1.3 channels41. In T-lymphocytes Charybdotoxin also blocked apoptosis, but Iberiotoxin, Apamin and Agitoxin failed to do so, ruling-out KCa1.1, KCa2 and KV1.3, strongly hinting at the involvement of KCa3.142.
Recently, a mechanism that links K+ efflux to the upstream apoptotic stimulus and mitochondrial disruption, was described in neurons46. It involves nitric oxide (NO) increase arising from the stimulus, leading to Zn2+ increase and activation of p38 MAPK. The latter activates K+ efflux directly, that again was found to be an obligatory step leading to cell death47.
It also should be mentioned that compensatory mechanisms might be activated. For example, KV1.3 was reported to mediate apoptosis but KV1.3 null mice thymocytes exhibited normal apoptosis that was dependent on Cl– channels47.
Plasma membrane Cl– and Na+ channels
Cl– channels must be active during cell volume changes to allow the net salt transport (NaCl or KCl) that is accompanied by water movement. In epithelial mammary cells, the expression of two Ca2+ activated Cl– channels is changed upon apoptosis induction. CLCA1, which is the dominant isoform under “normal” conditions, is down regulated while CLCA2 expression is upregulated48. The latter is in agreement with the disrupted expression of this channel in apoptosis resistant tumor cells. CLCA channels were also shown to be a necessary component in apoptosis of heptoma cell line, challenged with Cyclosporin A49. These observations point to the importance of CLCA channels (or their loss) in apoptosis and cancer.
Epithelial cells normally secrete Cl– alongside cations and a mutated Cl– Channel (CFTR) is the basis of a severe epithelial disease, cystic fibrosis (CF). CFTR channels were shown to support apoptosis in mouse mammary cells, where the natural mutant channel (causing the disease), failed to support such a process50. One role of the CFTR channel is to create an acidic environment, which allows the DNA of dying cells to be fragmented into small pieces. When the CFTR protein cannot fulfill this task, there is an accumulation of mucus with large DNA fragments. This may be the result of a process in which CF cells are not dying “properly” (epithelial tissue must renew constantly), due to a mutation in a channel that is needed for apoptosis. A similar mechanism was suggested in Jurkat T cells, where unidentified Cl– channels also mediate apoptosis and their block prevents cell acidification51.
Another, as yet unidentified, Cl– channel that is activated upon cell swelling plays a role in cardiac cell apoptosis during heart surgery and/or transplantation. However, there are contradicting reports regarding its role(s) as proapoptotic or protective channel. While administration of the Cl– channel blockers NPPB and IAA-94 seemed to enhance apoptosis in rabbit hearts52, NPPB and DIDS protected from apoptosis in rat53.
NaV54 and connexin (gap junction)55 channels were shown to control cell shrinkage and/or apoptosis in tumor cell lines, but the exact role played in apoptosis by these channels has not been extensively studied. However, Na+ inflow is necessary for Jurkat T cells induced shrinkage and conducting NaV channels are required for apoptosis as Saxitoxin (a NaV non-specific blocker), also blocked apoptosis54. The obligatory involvement of channels permeable to different ions in apoptosis suggests that the disruption of ion homeostasis is a necessary component of apoptosis propagation.
The Apoptosis-Cancer paradox of ion channels
One of the main motivations for trying to understand the role of ion channels in apoptosis is derived from a possible link to the role of the channels in cancer, where apoptosis is notoriously harder to achieve. As inbalance between apoptosis and proliferation may account for cancerous phenotypes, one may expect that K+channels, which must be upregulated in order for a cell to die, will be downregulated if the cell is to live forever.
In fact, this is not the case, and a few examples exist of certain K+ or Ca2+ channels that are upregulated in both cellular conditions. These include K2P9.1, KV1.3, KCa1.1 and P2X743,10,5. However, KV11.1 (HERG) channels were shown to play a dual role: in apoptosis as an active K+ channel and in proliferation as a membrane anchoring protein recruiting growth receptors to the membrane56. The duality of the KV11.1 effect was investigated with the use of a natural mutant. It lacks the ability to conduct ions and does not support apoptosis, but retains wild-type abilities to express in the membrane and bind the growth receptor. Specific antibodies used to co-immunopercipitate KV11.1 and TNF1R also showed this ability. This study is particularly interesting as it was conducted in different tumor cell lines, some which express the channel and tend to die upon apoptotic insult, while others do not express the channel and are resistant to the apoptotic insult56.
Taken together, the observations summarized here highlight the importance of ion channels in cellular mechanisms that control cellular integrity and fate. These roles played by ion channels in killing a cell are relevant in many clinical situations ranging from protection of the perfused heart during surgery, to exchanging proliferation for apoptosis as a possible tool in the fight against cancer.
References
- Bratton, S.B. and Cohen, G.M. (2001) Trends Pharmacol. Sci. 22, 306.
- Domen, J. (2001) J. Apoptosis 6, 239.
- Aartes, M. et al. (2003) Cell 115, 863.
- Sears, R.C. and Nevins, J.R. (2002) J. Biol. Chem. 277, 11617.
- Remillard, C.V. and Yuan, J.X.J. (2003) Am. J. Physiol. 286, L49.
- Matteson, M.P. and Chan, S.L. (2003) Nat. Cell Biol. 12, 1041.
- Nadeau, H. et al. (2000) J. Neurophysiol. 84, 1062.
- Li, H. et al. (2000) Science 289, 1159.
- Pal, S. et al. (2003) J. Neurosci. 23, 4798.
- Storey, N.M. et al. (2003) J. Biol. Chem. 278, 33319.
- Hille, B. (2001) Ion Channels in Excitable Membranes. 3rd edition.
- Zheng, L.M. et al. (1991) J. Cell. Biol. 112, 279.
- Fairbairn, I.P. et al. (2001) J. Immunol. 167, 3300.
- Humphreys, B.D. et al. (2000) J. Biol. Chem. 275, 26792.
- Gu, B. J. et al. (2001) J. Biol. Chem. 276, 11135.
- Coutinho-Silva, R. et al. (1999) Am. J. Physiol. 276, C1139.
- Nihei, O.K. et al. (2000) Blood 96, 996.
- Greig, A.V.H. et al. (2003) Journal of Investigative Dermatology 121, 1145.
- Chvatchko, Y. et al. (1996) Immunity 5, 275.
- Cavaliere, F. et al. (2002) J. Neurochem. 83, 1129.
- Cano-Abad, M.F. et al. (2001) J. Biol. Chem. 276, 39695.
- Juntti-Berggren, L. et al. (1993) Science 261, 86.
- Wang, L. et al. (1999) Endocrinology 140, 1200.
- Hara, Y. et al. (2002) Mol. Cell 9, 163.
- Zhang, W. et al. (2003) J. Biol. Chem. 278, 16222.
- Agopyan, N. et al. (2003) Am. J. Physiol. 286, L563.
- Zhang, L. et al. (2003) Cancer Gene Therapy. 10, 611.
- Boehning, D. et al. (2003) Nat. Cell Biol. 5, 1051.
- Jonas, E.A. et al. (2003) J. Neurosci., 23, 8423.
- Kirichok, Y. et al. (2004) Nature 427, 360.
- Crompton, M. (2000) J. Physiol. 529, 11.
- Degterev, A. et al. (2001) J. Cell. Biol. 155, 695.
- Shimizu, S. et al. (1999) Nature 399, 483.
- Cheng, E.H. Y. et al. (2003) Science 301, 513.
- Fernandez-Salas, E. et al. (2002) Mol. Cell Biol. 22, 3610.
- Suh, K.S. et al. (2003) J. Biol. Chem. 279, 4632.
- Teshima, Y. et al. (2003) Stroke 34, 1796.
- McCully, J.D. and Levitsky, S. (2003) Ann. Thorac. Surg. 75, S667.
- Xu, W. et al. (2002) Science 298, 1029.
- Yu, S.P. et al. (1997) Science 278, 114.
- Manikkam, M. et al. (2002) Biol. Reproduc. 67, 88.
- Elliot, J.I. and Higgins, C. F. (2003) EMBO Reports 4, 189.
- Lauritzen, I. et al. (2003) J. Biol. Chem. 278, 32068.
- Coroneo, M.T. et al. (2002) Invest. Ophthalmol. Vis. Sci. 43, E-Abstract 752.
- Trimarchi, J.R. et al. (2002) Am. J. Physiol. 282, C588.
- Bossy-Wetzel, E. et al. (2004) Neuron 41, 351.
- Koni, P. A. et al. (2003) J. Biol. Chem. 278, 39443.
- Elble, R.C. and Auli, B.U. (2001) J. Biol. Chem. 276, 40510.
- Kim, J. A. et al. (2003) Biochem. Biophys. Res. Commun. 309, 291.
- Gottlieb, R.A. and Dosanjh, A. (1996) Proc. Natl. Acad. Sci. USA. 93, 3587.
- Szabo, I. et al. (1998) Proc. Natl. Acad. Sci. USA. 95, 6169.
- Souktani, R. et al. (2003) Fundam. Clin. Pharmacol. 17, 555.
- Mizoguchi, K. et al. (2002) Transplantation. 73, 1185.
- Bortner, C.D. and Cidlowski, J.A. (2003) J. Biol. Chem. 278, 39176.
- Kalvelyte, K. et al. (2003) Biochem. Pharmacol. 66, 1661.
- Wang, H. et al. (2002) Cancer Res. 62, 4843.