Voltage-gated Ca2+ channels play a pivotal role in the regulation of a wide range of cellular processes, including membrane excitability, Ca2+ homeostasis, protein phosphorylation, gene regulation, muscle contraction and vesicular secretion. The L-type voltage-gated Ca2+ channels are the most common. This brief review aims at demonstrating advances made in research regarding voltage-gated Ca2+ channels in the cardiovascular system and the contributions of Alomone Labs in the field.
In cardiac and smooth muscle cells the predominant voltage-gated Ca2+ channels (CaV) are the dihydropyridine-sensitive CaV1.2 (α1C) channels (also called dihydropyridine receptor or L-type channel). These channels mediate long-lasting Ca2+ currents as a criticalstep in excitation-contraction coupling and have a central rolein controlling cardiac function2,3. The CaV1.2 channels provide a trigger for intracellular Ca2+ release during excitation-contraction coupling in a processknown as Ca2+-induced Ca2+ release. Dysregulationof Ca2+ channels in cardiac muscles leads to Ca2+ overload that influences the plateau phase of the cardiac action potential, and disrupts pacemaker activity in the sinoatrial node. The α1 subunit (the pore forming subunit) is a targetfor calcium channel blockers such as dihydropyridines (DHPs), phenylalkylamines, and benzothiazepines, which are widely used for treating hypertension, angina pectoris, and cardiac arrhythmias4,5.
In the cardiac muscle, a distinct α1 subunit (α11.2a cardiac isoform), an α2δ subunit, and several isoforms of β subunits (β1b and β2a-d) have been identified and implicated to form the CaV1.2 channel. The CaV1.2a α1C subunit is an alternative splice variant of exon 1 which differs from the vascular smooth muscle isoform at the N-terminus.
Two forms of the CaV1.2 α1C subunit (≈240 and 210 kDa) are expressed in the cardiac muscle which differ in a truncation at the C-terminus. Whereas the majority of α1C subunits isolated from cardiac muscle are truncated (demonstrated using Anti-CaV1.2a (CACNA1C) Antibody, (#ACC-013))6, the cleaved distal C-terminus remains associated with the truncated α1 subunit of CaV1.2a following proteolytic processing, and peptides derived from it can control the channel’s activity. The cardiac CaV1.2a activity is highly regulated by a variety of stimulus. The C-terminal region is a target site for β-Adrenergic regulation and for phosphorylation by PKA6. Stimulation of cardiac cells by Recombinant human LIF protein (human Leukemia Inhibitory Factor) (#L-200), enhances CaV1.2a currents through phosphorylation by ERK at S1829 of the α1C subunit6,7.
Although physiologically essential for normal cardiac function, increased level of CaV1.2 channels or activity have been implicated in pathological processesinvolving Ca2+ disturbance, including cardiac ischemia and myocardial stunning8. The expression level of CaV1.2 in the heart was demonstrated using Anti-CaV1.2 (CACNA1C) Antibody (#ACC-003) and its expression was up-regulated when PI3K and Ca2+/calmodulindependent protein kinase II (CamK II) were overexpressed9,10 (Figure 1).
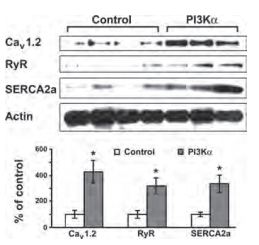
Western blot analysis (upper panel) and densitometric measurement (lower panel) of L-type Ca2+ channel protein expression in PI3-Kinase α inducible transgenic mice using Anti-CaV1.2 (CACNA1C) Antibody (#ACC-003). (Left: non-induced control mice, right: PI3-Kinase α induced mice).
Adapted from reference 9 with permission of The American Physiological Society.
In hypertension diseases, the level of vascular voltage-gated L-type calcium channel currents and vascular tone are increased, as a result of the CaV1.2 non-cardiac form overexpression (demonstrated in different rodent models of hypertension using Anti-CaV1.2 (CACNA1C) Antibody11.
The physiological importance of the β3 subunit in the cardiovascular system was investigated using gene targeted mutant mice. In this system the lack of β3 subunit in β3-nullmice was confirmed using Anti-CACNB3 Antibody (#ACC-008). Western blot analysis using Anti-CaV1.2a (CACNA1C) Antibody demonstrated a remarkable reduction in the membrane expression of α1C subunit in these mice. The reduction of α1C was accompanied by a significant decrease in Ca2+ current density, a slower inactivation rate of Ca2+ channel, and reduced dihydropyridine-sensitivity12.
CaV1.3 channels play a pivotal role in the generation of cardiac pacemaker activity by contributing to diastolic depolarization in the sinoatrial node (SAN). Abnormal CaV1.3 trafficking caused by ankyrin-B dysfunction leads to abnormal sinoatrial node (SAN) excitability and causes SAN disease. In this syndrome the cardiac distribution of CaV1.3 was demonstrated by immunocytochemistry staining using the Anti-CaV1.3 (CACNA1D) Antibody (#ACC-005) (Figure 2)13.
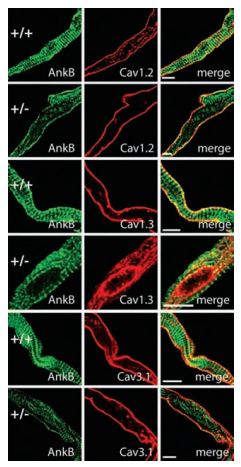
Confocal imaging of membrane distribution of voltage gated Ca2+channels in SAN cells of WT and Anykin B+/- transgenic mice was detected using Anti-CaV1.3 (CACNA1D) Antibody (#ACC-005), Anti-CaV1.2 (CACNA1C) Antibody (#ACC-003) and Anti-CACNA1G (CaV3.1) Antibody (#ACC-021).
Adapted from reference 13 with permission of The National Academy of Sciences of the USA (copyright 2008).
Ca2+ currents, through voltage-gated Ca2+ channels play a key role during excitation contraction coupling in smooth muscle cells (SMCs) of the vascular system. Using vascular smooth muscles cells, L-, P- and Q-type voltage-gated Ca2+ channels were found to be expressed using Anti-CACNA1A (CaV2.1) Antibody (#ACC-001) as well as Anti-CaV1.2 (CACNA1C) Antibody. Additional information about the identity of these channels was confirmed using Calciseptine (#SPC-500) known to inhibit the L-Type Ca2+ channels and ω-Agatoxin IVA (#STA-500) which inhibits P and Q-type Ca2+ channels14. Figure 3 shows smooth muscle cells treated with Calciseptine, showing that L-type Ca2+ channels are indeed involved Ca2+ currents in these cells15.

A) Depolarization-induced changes in [Ca2+]i in a smooth muscle cell in the vascular wall before, during, and after treatment with Calciseptine (#SPC-500) (100 nmol/l). B) Baseline [Ca2+]i in smooth muscle cells in untreated perfused arterioles (n=7), and arterioles treated with Calciseptine (100 nmol/l, n=5). C) KCl induced peak in [Ca2+]i in smooth muscle cells in perfused arterioles (n=7) and perfused arterioles treated with Calciseptine (100 nmol/l, n=5). Data are mean values ± SE. *P < 0.05.
Adapted from reference 15 with permission of The American Physiological Society.
References
- Foell, J.D. et al. (2004) Physiol. Genomics 17, 183.
- Reuter, H. (1979) Annu. Rev. Physiol. 41, 413.
- Catterall, W.A. (2000) Annu. Rev. Cell Dev. Biol. 16, 521.
- Reuter, H. (1967) J. Physiol. 192, 479.
- Bers, D.M. (2002) Nature 415, 198.
- Ganesan, A.N. et al. (2006) Circ. Res. 98. e11.
- Takahashi E. et al. (2004) Circ. Res. 94, 1242.
- Bolli, R. and Marban, E. (1999) Physiol. Rev. 79, 609.
- Yano, N. et al. (2008) Am. J. Physiol. Heart Circ Physiol. 295, 1690.
- Wang, Y. et al. (2008) J. Biol. Chem. 283, 25524.
- Pratt, P.F. et al. (2002) Hypertension 40, 214.
- Murakami, M. et al. (2003) J. Biol. Chem. 278, 43261.
- Le Scouarnec, S. et al. (2008) PNAS 105, 15617.
- Andreasen, D. et al. (2006) Hypertension. 47, 735.
- Uhrenholt, T.R. et al. (2007) Am. J. Physiol. Renal Physiol. 292, F1124.