Venomous creatures deploy a wide array of proteins and peptides, active on a spectrum of targets, for prey immobilization. These toxins are small, rigid, highly potent and specific, and as such, serve as an important pharmacological tool for ion channel research. Some of these toxins are currently under evaluation as possible drugs for a number of pathological indications.
Introduction
Venomous creatures deploy a wide arsenal of biologically active compounds to capture prey, acting on a vast array of targets.1 Prey immobilization is achieved by either inhibiting or activating the electrical activity using very specific and potent compounds. The diverse array of venom components belong to a number of compound classes, including small molecular weight compounds, electrolytes, polyamines, neurotransmitters, amino acids, small peptides and high MW proteins. Recently it was reported that a single type of spider venom might contain up to 500 peptides and therefore, with an estimated 37,000 known species, it would represent a potential source of more than 18 million spider peptides.1,2 The great pharmacological wealth accumulated by nature over evolution has resulted in extremely complex mixtures of pharmacophores, a combinatorial library of hundreds of thousands of potentially active and useful molecules.3 Venom components target different cellular mechanisms, and in many species, they specifically target ion channel families, which are the major components responsible for cellular membrane excitability.4 Due to their high potency and selectivity, venom proteins and peptides have emerged as invaluable tools for research, drug discovery and drug development.5,6 A number of venom toxins have been approved by the FDA as therapeutics, while others are currently in various stages of clinical trials as potential drugs for a number of indications, including pain, cardiovascular, and neurological pathologies.5,6 Due to the huge scope of this subject, this review we will highlight only a small fraction of examples for the use of venom toxins for the pharmacological characterization of ion channels.
Mode of Action of Toxins and the Structure of Ion Channels
Complex multicellular organisms require rapid and accurate transmission of information among cells and tissues and tight coordination of distant functions. Ion channels are integral membrane proteins that span lipid bilayers to form a central pore through which selected ions can pass at near diffusion-limited rates (approximately 107 ions-1 s-1).4 They are found in a wide range of organisms from viruses and bacteria to plants and animals.4,7,8 Most ion channels are gated, capable of making transitions between conducting and non-conducting conformations. Channel gating can be induced by extracellular ligands, intracellular second messengers and metabolites, protein–protein interactions, phosphorylation, and other factors.4,7 In addition, many ion channels are gated by the membrane potential itself.4 Voltage-gated ion channels respond to and modify the changes in membrane potential. For example, in vertebrates electrical signals and the resulting intracellular Ca2+ transients control contraction of muscle, secretion of hormones, perception of the environment, processing of information in the brain, and output from the brain to peripheral tissues.4 In nonexcitable cells, Ca2+ transients signal many key cellular events, including secretion, gene expression, and cell division.
The architectural structure of the ion channel families consists of four variations built on a common pore-forming structural theme.7,8 Voltage-gated K+ channels exemplify the first basic theme in the ion channel superfamily.4 This basic structure consists of six regions that form membrane-spanning helices (termed segments S1 to S6) and a membrane-spanning loop between the S5 and S6 segments (Fig. 1). Other members of this superfamily are the voltage-gated Na+ and Ca2+ channels.7 Their principal subunits are composed of four homologous KV domains (I to IV) that form the common structural motif for this family (Fig. 1).7-9 Experimental analysis of the ion channel structures shows that the four homologous domains surround a central pore and suggests laterally oriented entry ports in each domain for ion transit toward the central pore.4 Several other families of ion channels also have this tetrameric architecture. The inwardly rectifying K+ channels constitute the simplest structural motif in the ion channel protein superfamily. These channels are complexes of four subunits that each have only two transmembrane segments, termed M1 and M2, which are analogous in structure and function to the S5 and S6 segments of voltage-gated Na+, Ca2+, and K+ channels. Two of these pore motifs are linked together to generate the fourth structural theme, the two-pore K+ channels (Fig. 1). In the last few years, this field has advanced due to the solution of the three-dimensional structure of a number of K+-channels, including KcsA, KvAP and Kv1.2 by Roderick MacKinnon’s group, for which Prof. MacKinnon was awarded the 2003 Nobel Prize in chemistry.10-12
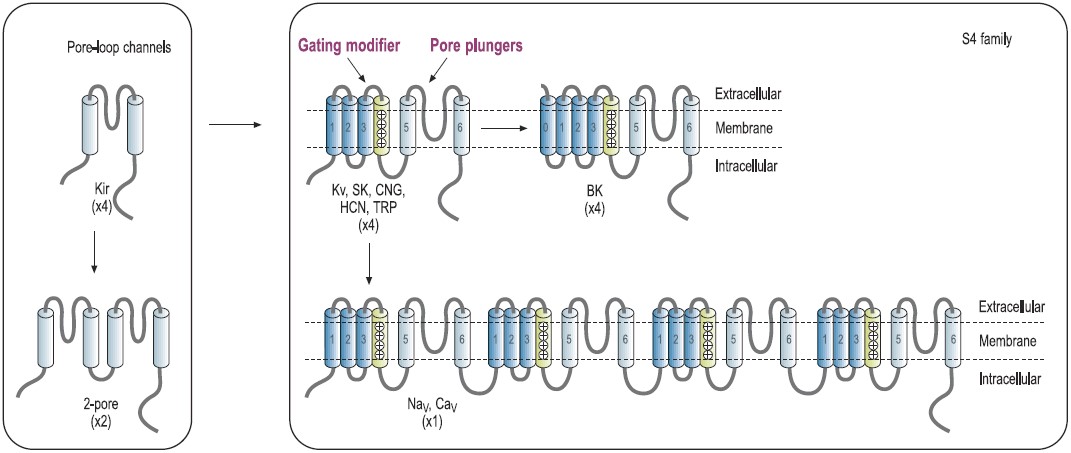
Predicted transmembrane topology of different types of channels. The numbers in parentheses refer to the number of subunits that make up the channel pore. The boxes indicate ligand-binding domains. The S4 domain is marked. KV, voltage-gated K+ channel. NaV, voltage-gated Na+ channel. CaV, voltage-gated Ca+ channel. CNG, cyclic-nucleotide-gated channel. HCN, hyperpolarization-activated channel. TRP, transient receptor potential channel.
Much of the knowledge on channel structure and function was elucidated using specific peptide ligands isolated from a number of venomous organisms. Toxins isolated from venomous animals are usually small peptides, ranging from 8-70 amino acids, with relatively small scaffold structures. They are highly compact and are mainly stabilized by disulfide bonds, although some are stabilized by hydrogen bonds made from post-translational-modified amino acids.6 Toxins have various pharmacological advantages including high specificity, high activity, no accumulation in organs, low toxicity and low immunogenicity. Furthermore, their small, disulfide cross-linked compact structures lead to enhanced chemical and thermal stability and relative insensitivity to proteases.13-15 The mode of action of these toxins can be roughly divided into two major groups: The “pore-plungers” and the “voltage-sensor modulators” or “gating-modifiers”. “Pore-plungers” bind to the pore region and physically block the ion transport through the pore. For example, the majority of K+ channel blockers are characterized by the presence of a dyad or triad, having a Lys in structural proximity to either one or two hydrophobic residues (Phe, Tyr, Trp).16 The Lys residue is “plunged” into the pore and interacts with the channel’s Asp residues inside the pore, while the hydrophobic residues of the toxin interact with hydrophobic residues on the pore outer-surface.17 Toxin-channel selectivity is achieved by additional protein-protein interactions, thus discriminating between closely related channels. “Gating-modifier” toxins bind to the voltage sensor moiety (S4 helix), which is partially exposed to the extracellular space, modulates its movement, and thus modulates the mechanism of pore gating, although the exact mechanism remains controversial.12,18,19 Since these scaffolds are gaining interest as possible drugs for a vast array of indications, much research (including mutagenesis experiments, in-silico simulations, and structure determinations20-25) has been invested into elucidation of their structure/function relationship.
Scaffold Structures of Venom Peptides and Proteins
The majority of the experimental data acquired to date has been from toxins isolated from the venoms of snakes, scorpions, spiders, marine snails (Conus genus) and sea anemones.1,26-28 The use of toxin scaffolds as therapeutics has been the subject of a number of reviews.5,6,29-30
Although a substantial number of toxin structures have been resolved to date, they represent a minute amount of all possible scaffold structures and activities of toxins in venoms. Classification into families and subfamilies is performed by structural and sequence data. Nevertheless, evolutional diversity is found, with at least 14 scaffold representatives characterized.13 The scaffolds are composed of a combination of α-helices (α) and β-sheets (β), and can be roughly categorized as α310, α, αβ, αα, 310α, β, ββ, βββ, β1β1β2β2β2, αββ, βαββ, α/β scaffolds (for clarification of the scaffolds, please refer to Mouhat, S. et al.13). A number of representative scaffolds are illustrated in (Fig. 2). At least eight distinct toxin scaffolds have been identified for K+ channel inhibition, eight scaffolds for Na+ channel modulation and four for Ca2+ channel blocking activity, where overlapping scaffolds are found between channel subtypes. As an example, the βββ (Inhibitory Cysteine Knot) scaffold is found in κ-PVIIA (Conus purpurascens) which blocks KV channel, ACTX-Hi:OB4219 (from the spider Hadronyche infensa) which slows the inactivation of NaV channels, and ω-Conotoxin GVIA (#C-300) (marine cone snail Conus geographus) which blocks CaV channel activity.13 Furthermore, each of the scaffolds can be further divided into subgroups, for example, at least 18 sub-groups can be identified (based on Cys-pairing and activity) in the K+-channel blocker α/β α-KTX scorpion toxins family.31 It is fascinating to observe that the same channel can be blocked by a number of scaffolds that, in some cases, share a similar diad or triad, but in other cases, block the channel by a different mechanism, as exemplified by Tc32 (isolated from the venom of the Amazonian scorpion Tityus cambridgei).16, 32 Alternatively, similar toxins sharing the same scaffold can have very distinct ion channel targets, for example the spider toxin Stromatoxin-1 (#STS-350) (Stromatopelma calceata) is active on the K+-channel KV2.1 while SNX-482 (#RTS-500) (Hysterocrates gigas) is active on the R-type Ca2+ channel (CaV2.3).13 Additionally, APETx1 and APETx2, isolated from the sea anemone Anthopleura elegantissima, share 65% sequence identity and 76% homology, but are active on very distinct targets namely, KV11.1 and ASIC3, respectively.
For an extensive treatment of this topic see Refs. 13-15.
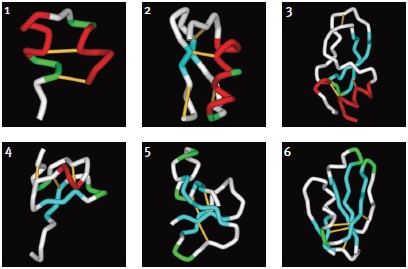
1. κ-Hefutoxin-1: αα, (1HP9). Two consecutive α-helices within a peptide or protein structure.
2. Maurotoxin (#STM-340): αββ, (1TXM). Combination of α-helix (N-terminal) followed by a two-stranded β-sheet (C-terminal).
3. Dendrotoxin-1 (#D-390): 310ββα(1DEM). Kunitz/Bovine pancreatic trypsin inhibitor, arranged to form a twisted two-stranded anti-parallel β-sheet followed by an α-helix.
4. Stromatoxin-1 (#STS-350): 310ββ(ICK) (based on the related structures of Hanatoxin-1 and ω-Grammotoxin SIA (#G-450), 1D1H.pdb and 1KOZ.pdb, respectively)46. Belongs to the Knottin superfamily, having the Inhibitory Cysteine Knot structure, achieved when one disulfide bridge crosses the macrocycle formed by the two other disulfides and the interconnecting backbone.
5. ATX-II (#STA-700): ββββ (1ATX), belongs to the Defensin/myotoxin-like superfamily. All β structures are crosslinked by 3 cysteine bridges.
6. FS-2 (#F-700): β1β1β2β2β2 (1TFS). Three-finger motif, two consecutive (1 and 2) β-sheets (anti-parallel) within a peptide or protein structure (the first one being a two-stranded β-sheet and the second one a three-stranded β-sheet).Images were generated using the ViewerLite freeware (Accelrys Inc.). The model structure of Stromatoxin was generated using Swiss Model.
Use of Toxins as Pharmacological Tools
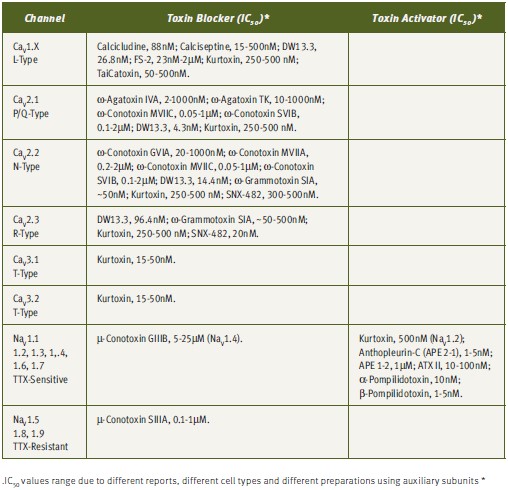
The understanding of the biophysical and pharmacological properties of channels has been greatly improved by the use of toxins, particularly in differentiation of specific channel activities (see Tables 1 and 2). The electrical currents generated by different channels can now be modulated by the use of toxins. As has been mentioned earlier, although numerous toxins have been discovered, they represent only a minute fraction of the toxin repertoire present in venoms.2,28 As more toxins are discovered, more channels and channel subtypes can be characterized. An example of differentiation of channel type by the use of toxins is given by the Ca2+ channels. Dihydropyridine-sensitive L-type Ca2+ channels (CaV1.x) can be selectively blocked by a number of toxins, including, Calcicludine (#C-650, replaced with #SPC-650) (Dendroaspis angusticeps), Calciseptine (#C-500), FS-2 (#F-700) (Dendroaspis p. polylepis) and TaiCatoxin (Oxyuranus s. scuttlelatus), while high voltage-activated dihydropyridine-insensitive (CaV2.x) channels can be differentiated using the toxins ω-Agatoxin IVA (#STA-500) (Agelenopis aperta) for P/Q-type (CaV2.1), ω-Conotoxin GVIA (#C-300) for N-type (CaV2.2) and SNX-482 (#RTS-500) for R-type (CaV2.3) Ca2+ channels. Low voltage-activated Ca2+-channels (CaV3.x) can be blocked by the relatively T-type selective blocker Kurtoxin (Parabuthus transvaalicus).33-35 Furthermore, it was recently published that even N-type channel variants, arising from splice isoforms or differential modulation by auxiliary subunits, can be distinguished using ω-Conotoxin-CVID (Conus catus), currently in clinical trials for pain indications (Table 2).36-37

Toxins have been shown to discriminate between members of the Ca2+-activated K+ channel family. Large-conductance Ca2+-activated K+ channel (BK, KCa1.1) is blocked by Iberiotoxin (#STI-400) (Mesobuthus tamulus), Slotoxin (#STS-410) (Centruroides noxius), Noxiustoxin (#STN-340) (Centruroides noxius), Kaliotoxin (#STK-370) (Androctonus m. mauritanicus), and Charybdotoxin (#STC-325) while the small-conductance Ca2+– activated K+ channel (KCa2x) can be differentially blocked using Apamin (#STA-200) (Apis mellifera), Scyllatoxin (#STS-370) (Leiurus quinquestriatus hebraeus) and Tamapin (#STT-400) (Mesobuthus tamulus). Intermediate-conductance Ca2+-activated K+ channel (IK, KCa3.1) can be selectively modulated by Maurotoxin (#STM-340) (Scorpio maurus palmatus) and Charybdotoxin (#STC-325) (Leiurus quinquestriatus hebraeus) (Table 1).38
Other examples of unique targets blocked by toxins can be illustrated by KV11.1 (HERG) K+ channel block using Ergtoxin-1 (#STE-450) (Centruroides noxius),39 BeKm-1 (#STB-470) (Mesobuthus eupeus)40 and APETx1 (Anthopleura elegantissima)41, ASIC1a channel using Psalmotoxin42, ASIC3 channel using APETx2 (#STA-160) (Anthopleura elegantissima)43, Kir3.1 (GIRK) using Tertiapin-Q (Apis mellifera)44, and Kir1.1 (ROMK1) using Lq2 (#RTL-550) (Leiurus quinquestriatus hebraeus).45
Due to their small size, compact and rigid structure, high potency and selectivity, toxins have emerged as highly valuable tools for research and drug development for numerous indications. Tables 1 and 2 summarizes the published IC50 for a number of toxins against their targets, which emphasizes the strength of these reagents as excellent pharmacological tools.
References
- Escoubas, P. (2006) Toxicon 47, 609.
- Escoubas, P. et al. (2006) Toxicon 47, 650.
- Theakston, R.D. and Kamiguti, A.S. (2002) Toxicon 40, 579.
- Hille, B. (2001) Ion Channels in Excitable Membranes. 3rd edition.
- Bogin, O. Venom Peptides and their Mimetics as Potential Drugs, Modulator Issue #19.
- Lewis, R.J. and Garcia M.L. (2003) Nat. Rev. Drug. Discov. 2, 790.
- Yu, F.H and Catterall, W.A. (2004) Sci. STKE. 253, re15.
- Ashcroft F.M. (2006) Nature 440, 440.
- Shieh, C.C. et al. (2000) Pharmacol. Rev. 52, 557.
- Doyle, D.A. et al. (1998) Science 280, 69.
- Lee, S.Y. et al. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 15441.
- Long, S.B. et al. (2005) Science 309, 897.
- Mouhat, S. et al. (2004) Biochem. J. 378, 717.
- Rodriguez de la Vega, R.C. et al. (2005) Toxicon 46, 831.
- Cheek, S. et al. (2006) J. Mol. Biol. 359, 215.
- Mouhat, S. et al. (2005) J. Pept. Sci. 11, 65.
- Rodrıguez de la Vega, R.C. et al. (2003) Trends Pharmacol. Sci. 24, 222.
- Phillips, L.R. et al. (2005) Nature 436, 857.
- Jung, H.J. et al. (2005) Biochemistry 44, 6015.
- Gilquin, B. et al. (2005) J. Biol. Chem. 280, 27093.
- Cestele, S. et al. (2006) J. Biol. Chem. M603814200.
- Wu, Y. et al. (2004) Biophys. J. 87, 105.
- Andreotti, N. et al. (2005) Peptides 26, 1095.
- Korolkova, Y.V. et al. (2004) J. Mol. Recognit. 17, 209.
- Visan, V. et al. (2006) Mol. Pharmacol. 66, 1103.
- Rash, L.D. and Hodgson, W.C. (2002), Toxicon 40, 225.
- Honma, T. and Shiomi, K. (2006) Mar. Biotechnol. (NY), 8, 1.
- Terlau, H. and Olivera, B.M. (2004) Physiol. Rev. 84, 41.
- Ménez, A. (1998) Toxicon 36, 1557.
- Gilquin, B. et al. (2003) Protein Sci. 12, 266.
- Rodriguez de la Vega, R.C. and Possani, L.D. (2004) Toxicon 43, 865.
- Batista, C.V. et al. (2002) Biochim. Biophys. Acta. 1601, 123.
- Chuang, R.S. et al. (1998) Nat. Neurosci. 1, 668.
- Sidach, S.S. and Mintz, I.M. (2002) J. Neurosci. 22, 2023.
- Meir, A. (2005) Voltage Dependent Ca2+ (CaV) Channels, Modulator Issue #20.
- Lewis, R.J. et al. (2000) J. Biol. Chem. 275, 35335.
- Smith, M.T. et al. (2002) Pain 96, 119.
- Bronstein-Sitton, N. (2005) Ca2+-dependent K+ (KCa) channels: At the Crossroads of Cell Metabolism, Modulator Issue #20.
- Scaloni, A. et al. (2000) FEBS Letters, 479, 156.
- Korolkova, Y.V. et al. (2001) J. Biol. Chem. 276, 9868.
- Diochot, S. et al. (2003) Mol. Pharmacol. 64, 59.
- Escoubas, P. et al. (2003) Protein Sci. 12, 1332.
- Diochot, S. et al. (2004) EMBO J. 23, 1516.
- Jin, W. and Lu, Z. (1998) Biochemistry 37, 13291.
- Lu, Z. and MacKinnon, R. (1997) Biochemistry 36, 6936.
- Peitsch, M.C. (1996) Biochem. Soc. Trans. 24, 274.