N-Methyl-D-Aspartate Receptor (NMDAR) are ubiquitously expressed along the central nervous system and are instrumental to various physiological processes such as synaptic plasticity and learning. Nevertheless, several mental disabilities including schizophrenia and Alzheimer’s disease are all related to NMDAR dysfunction. Here, we review many aspects of NMDAR function and regulation and describe their involvement in pathophysiological states using Alomone Labs products.
Introduction
Glutamate is a key neuro-transmitter in the central nervous system and acts on a variety of cell surface receptors, collectively termed ionotropic glutamate receptors (iGluRs)15. The N-Methyl-D-Aspartate receptors (NMDAR) are members of the iGluR superfamily and are pivotal to many physiological processes such as the formation of long term memory, synaptic plasticity and many other cognitive functions. Therefore, it is not surprising that several mental disorders including schizophrenia, epileptic aphasia and other debilitating neurodegenerative diseases such as Alzheimer’s are all related to NMDAR dysfunction21.
Structure
Most NMDAR proteins form a functional heterotetramer protein-complexe by incorporating the obligatory NR1 (GluN1) subunit with different types of the four NR2 subunits, termed NR2A-D (GluN2A-D) or two NR3 isoforms (GluN3A,B)6, 19. Furthermore, this heterogenic complexity is further increased by the fact that NR1 is predisposed to alternative splicing events and can give rise to additional eight proteins19. Eventually, this diversity of protein assemblies brings about a variety of NMDAR receptors with different biophysical properties and expression patterns throughout the nervous system15.
Each of these NMDAR protein complexes contain an extracellular N-terminal domain and a ligand binding domain for glycine on GluN1 and glutamate binding domain on GluN2 and GluN3 subunits6. In addition, the intracellular carboxy tail is another important domain as it has an impact on receptor trafficking, anchoring and mediates various interactions with intracellular signaling proteins15.
Mechanism of Action
NMDAR activation depends on sequential conformational changes to relieve the magnesium blockade which is achieved by rapid membrane depolarization and binding of both glycine and glutamate ligands6, 21. This in turn removes the inhibitory electrostatic forces of magnesium and enables calcium influx and transmission of long lasting signals (i.e. long-term potentiation), a key mechanism to learning and memory formation10.
Regulation
Given their large diversity and wide-spread expression across the nervous system and the fact that NMDARs engage in essential physiological processes related to cognitive performance, it is not surprising that over the last decade numerous efforts were invested in understanding the regulation and function of these receptors.
NMDAR Function and Turnover by Scaffold Proteins
Neuronal communication occurs through synaptic connections where a presynaptic dendrite transmits a signal towards the postsynaptic body of another neuron. NMDARs are critical for these neuronal circuits and are tethered in the post synaptic density (PSD) areas by forming numerous connections with scaffold proteins and other cell-signaling mediators10. Homer proteins are an example of PSD scaffold proteins that mediate the connection between group-I metabotropic glutamate receptor subtype 5 (mGluR5) and NMDAR. Recently, Aloisi et al., investigated the consequences of Homer disruption in the context of fragile x syndrome, where intellectual disabilities are frequently associated with abnormal mGluR5 and NMDAR functions1. Using Anti-NMDAR1 (GluN1) (extracellular) Antibody (#AGC-001), the authors uncovered a novel mechanism through which mGluR5 suppresses NMDAR activity in hippocampal neurons obtained from Fmr1 knockout (KO) mice. Quantum dot tracing demonstrated that GluN1 and mGluR5 cluster in the synaptic area, suggesting a possible existence of a physical interaction1. Indeed, confocal microscopy showed co-localization of GluN1 with mGluR5, which increased in Fmr1 KO neurons1 (Figure 1). Lastly, the authors demonstrated that the mGluR5-NMDAR interaction attenuates NMDAR signal transduction as measured by NMDAR-dependent excitatory post-synaptic currents (ESPC).
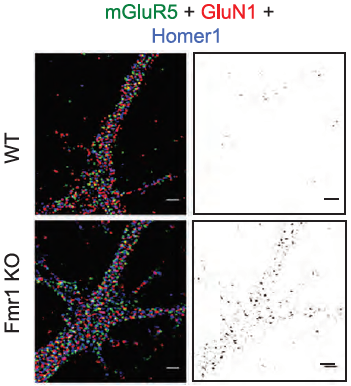
.Immunocytochemical staining of mouse hippocampal neurons using Anti-NMDAR1 (GluN1) (extracellular) Antibody (#AGC-001), (red). Triple immunostaining experiment indicates increased synaptic clustering of GluN1, mGluR5 and Homer1 in Fmr1 knockout mice.
Adapted from reference 1 with permission of SPRINGER NATURE.
PSD-95 is another important scaffold protein that mediates NMDAR anchoring at the PSD membranes of postsynaptic neurons19. PSD-95 activity is regulated by different mechanisms including phosphorylation on serine/ threonine residues by the peptidyl-prolyl cis-/trans isomerase, Pin12. This phosphorylation event induces conformational changes in PSD-95 and determines its substrate specificity2. The effect of PSD-95 interaction with NMDAR via its phosphorylation by Pin1 was investigated2. Anti-NMDAR2B (GluN2B) (extracellular) Antibody (#AGC-003) was used to probe GluN2B expression after immuno-purification of PSD-95 from brain extracts of Pin1 KO mice or their wild type littermates2. GluN2B expression was significantly increased in brain lysates from Pin1 KO compared to wild type2. Similarly, over-expression of GluN2B was also observed in hippocampal protein extracts of Pin1 KO mice by western blotting2. Taken together, these data place Pin1 as a negative modulator of PSD-95-NMDAR interaction.
In light of the prominent role of scaffold proteins for neuronal cell function, Scribble1 (Scrib1) was found to be a critical modulator of NMDARs spatial distribution16. In particular, Scribble1-NMDAR interaction protects NDMARs from lysosomal degradation and thus, increases their synaptic cell-surface concentration. Investigation into the mechanisms that underpins Scribble1- NMDAR interactions, zeroed in on the PDZ domain of Scribble1 as the site of protein interaction16. Green fluorescent protein (GFP), Scribble1-GFP and a Scribble-mutant isoform, lacking two PDZ domains were overexpressed in primary hippocampal neurons and cell-surface levels of NMDAR were determined by fluorescent microscopy, using Anti-NMDAR2B (GluN2B) (extracellular) Antibody16. For quality control purposes, Anti-GABA(A) α1 Receptor (extracellular) Antibody (#AGA-001) was used to show that ectopic expression of Scrib1 did not alter the nature of these neurons. Thus, increased cell-surface levels of NMDAR due to Scribble interaction are specific and depend on PDZ2 and PDZ3 domains16.
Regulation of Synaptic NMDAR Composition by Protein Degradation
Synaptic NMDAR composition is a highly dynamic process that has a substantial impact on signal transduction and developmental processes15. Synaptic composition is regulated by different mechanisms including transcriptional networks, protein trafficking systems but also through active proteolytic degradation10, 19. Accordingly, the ubiquitin ligase, F-box Only Protein 2 (Fbxo2) regulates synaptic expression of GluN1-GluN2A subunits by means of selective subunit degradation. Given the fact that Fbxo2 promotes GluN1 degradation is established, an in vitro model of non-neuronal cells was used to compare the effect of Fbxo2 on GluN2A and GluN2B using the specific Anti-NMDAR2A (GluN2A) (extracellular) Antibody (#AGC-002) and Anti-NMDAR2B (GluN2B) (extracellular) Antibody3. Intriguingly, a negligible effect of Fbxo2 on GluN2B degradation was observed as evident by western blotting. Likewise, brain lysates from Fbxo2 KO mice displayed higher levels of GluN2A, while GluN2B expression remained unchanged3. To examine these observations from a different angle, Atkin et al., used the above-mentioned antibodies in immunocytochemical staining of primary hippocampal neurons. As anticipated, GluN2A expression was greater in Fbxo2 KO neurons compared to wild type neurons, while GluN2B levels seemed to be down-regulated (Figure 2)3. Finally, the authors devised an ELISA assay, using Anti-NMDAR1 (GluN1) (extracellular) Antibody from Alomone Labs to verify whether increased synaptic expression of GluN1- GluN2A is due to decreased protein internalization. Indeed, cell surface expression of GluN1 was higher in Fbxo2 KO neurons compared to control after treatment with bicuculline (to stimulate NMDAR internalization)3. Overall, these data shed light on a new model for neuronal plasticity, in which synaptic NMDAR composition is regulated by selective protein degradation.
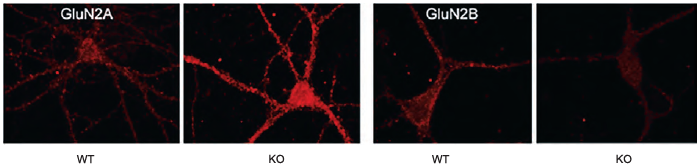
Immunocytochemical staining of mouse hippocampal neurons using Anti-NMDAR2A (GluN2A) (extracellular) Antibody (#AGC-002) and Anti-NMDAR2B (GluN2B) (extracellular) Antibody (#AGC-003). GluN2A staining (left) increases in Fbxo2-/- neurons while GluN2B staining (right) decreases.
Adapted from reference 3 with permission of the Society for Neuroscience.
Synaptic NMDAR Trafficking and Subunit Dynamics
Lateral mobility shifts of NMDARs across the plasma membrane, are important for enabling neurons to rapidly respond and adapt to various environmental stimuli. In particular, enrichment of synaptic GluN2A over GluN2B containing NMDAR can have a large impact on synaptic long-term potentiation (LTP) however, the mechanisms underlying this remodeling process are not fully understood. Dupuis et al., have recently studied this process by tracing GluN2A and GluN2B mobility via single molecule quantum dots assay4. To do so, the authors used Anti-NMDAR2A (GluN2A) (extracellular) and Anti-NMDAR2B (GluN2B) (extracellular) antibodies in primary hippocampal neurons followed by chemical stimuli to generate neuronal LTP. GluN2B rapidly diffused to the peri-synaptic area, while GluN2A remained relatively stable in the PSD region4. In an antibody cross-linking experiment to immobilize GluN2B using the Alomone Labs GluN2B or GluN1 antibodies in cultured neurons, the authors noted that LTP signals were markedly suppressed4. Moreover, these observations were fully recapitulated in rat hippocampal brain slices following GluN1 cross-linking. Together these data imply on a causal relationship between NMDAR dynamics and perpetuated signal transmission. To delve deeper into the mechanism that underlies NMDAR dynamics, the authors tested several inhibitors and monitored GluN2B dynamics by using Anti-NMDAR2B (GluN2B) (extracellular) Antibody coupled to quantum dot particles4. Data show that chemical inhibition or genetic manipulation of Ca2+/calmodulin-dependent protein kinase II (CaMKII), largely reduced GluN2B-NMDAR membrane dynamics4. Altogether, Dupuis et al., provide a mechanism through which CaMKII- GluN2B interaction reshapes synaptic NMDAR in response to stimuli.
Unlike CaMKII, which seems to support learning and memory, the protease tissue type plasminogen activator (tPA) seems to do the opposite. Lesept et al., provide fresh insights into the mechanism through which, tPA-NMDAR might be related to cognitive decline and neurotoxicity11. Using Anti-NMDAR1 (GluN1) (extracellular) Antibody in quantum dot application, they show that exogenous tPA increased NMDAR trafficking in extra-synaptic regions of cultured hippocampal neurons11. Furthermore, this diffusion pattern was mediated by tPA, as mutated tPA-protein did not replicate this mobility pattern11. Since tPA-NMDAR interaction increased calcium influxes in cultured neurons, the authors set out to investigate the influence of NMDAR mobility- shifts on intracellular calcium fluxes11. For this purpose, they decreased NMDAR surface mobility by antibody cross-linking. Consequently, receptor cross-linking generated extra-synaptic NMDAR clusters and increased intracellular calcium influx in response to stimuli11, suggesting that tPA increased extra-synaptic calcium signaling owing to NMDAR clusters11. Using quantum dot tracing, the authors showed that tPA-NMDAR interaction stimulated NMDAR mobility in extra-synaptic regions and provided evidence for this mechanism to be involved in neurotoxicity and cell death.
In addition to protein-protein interactions, synaptic plasticity is also regulated by external factors such as hormones. Stress hormones like corticosteroids are rapidly released in response to environmental stress and regulate synaptic plasticity through various mechanisms including transcriptional regulation, but not limited to12. Mikasova et al., discovered a non-genomic mechanism by which corticosteroids trigger full-blown excitatory signals by regulating synaptic NMDAR dynamics12. Specifically, using Anti-NMDAR1 (GluN1) (extracellular) Antibody to block NMDAR mobility in cultured hippocampal neurons, the authors uncovered that synaptic signal potentiation due corticosterone necessitates synaptic remodeling of NMDAR12.
Given that NMDAR dynamics is key to LTP signals and cognitive performance, it is likely that several brain disorders such as schizophrenia (SCZ) and encephalitis are related to impaired synaptic NMDAR remodeling. Jézéquel et al., demonstrated the presence of autoantibodies against NMDAR in patients with schizophrenia, but also in a small subgroup of healthy population by co-immunostainings of GluN1 expressed in embryonic kidney cells (HEK-293T) using the Alomone Labs antibody and IgGs purified from different donors (Figure 3)7. In addition, they also used brain hippocampal slices to validate their in vitro findings7. To reaffirm that patient-derived IgGs target NMDAR, the single nanoparticle quantum dots method was used to assess NMDAR membrane dynamics, given that different membrane proteins can be distinguished owing to different biophysical properties7. To demonstrate this, the authors labeled GluN1 using Anti-NMDAR1 (GluN1) (extracellular) Antibody as a positive control, patient-derived IgGs and an Anti-KV1.3 Antibody from Alomone Labs, as a negative control and monitored their diffusion rates using quantum dots in live hippocampal neurons7. As anticipated, all targets displayed the same diffusion pattern, except for the potassium channel7. This confirms the presence of GluN1 autoantibodies in patients with SCZ as well as in a minor group of healthy population. To get a better understanding on how NMDAR-directed autoantibodies affect synaptic remodeling, the authors quantified the content of synaptic NMDAR in hippocampal neurons by confocal microscopy. Collectively, these data highlight the inflammatory basis of SCZ and encephalitis and suggest a mechanism by which autoantibody production affects neuronal synapses7.
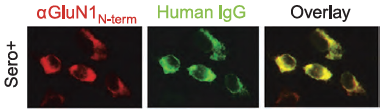
Immunocytochemical staining of live intact transfected HEK293 cells. Extracellular staining of cells with Anti-NMDA Receptor 1 (GluN1) (extracellular) Antibody (#AGC-001), (red), (left panel). Purified Human NMDAR IgGs (green) stain the transfected cells, (middle panel). Merge panel (right) shows complete overlap between the two stainings.
Adapted from reference 7 with permission of SPRINGER NATURE.
Given the inflammatory nature of several psychiatric disorders and the presence of autoantibodies for NMDAR in these patients, the provocative speculation for a possible immunotherapy to improve the outcomes in these patients was raised8. However, current diagnostic tools suffer from a lack of sensitivity when encountering patients with low antibody titers8. To work around this limitation, Jézéquel et al., devised a sensitive bioassay based on quantum dot tracing of NMDAR using the GluN1 antibody from Alomone Labs in hippocampal neurons. In particular, the authors demonstrated that diffusion trajectories from patients with low titer antibodies are similar to those from patients with high titer antibodies8. Thus, quantum dot tracing outperforms the standard diagnostic assays as these patients would most probably be erroneously classified as false negatives.
NMDARs in Development and Developmental Disorders
Cortical neurons display a remarkable decline in functional plasticity during adulthood which is associated with two main events: the formation of hyaluronic acid-based extracellular matrix (ECM) and the preferential expression of GluN2A over GluN2B containing NMDAR17. Whether these two biological processes are related remains currently unknown17. A study by Schweitzer et al., examined the influence of hyaluronic acid-based ECM on GluN2A and GluN2B expression in cortical neurons using Anti-NMDAR2A (GluN2A) (extracellular) Antibody and Anti-NMDAR2B (GluN2B) (extracellular) Antibody. Surprisingly, when cultured hippocampal neurons were treated with Hyaluronidase (Hya) to remove hyaluronic acid-based ECM, no difference in GluN2B expression was observed as evident by western blot17. However, confocal microscopy studies on living hippocampal neurons showed that surface expression of GluN2B increased following Hya treatment (Figure 4)17. Furthermore, this phenomenon was not confined to the synapses but was also evident in extra-synaptic regions implying that this reflects a global effect rather than local synaptic plasticity. The fact that surface expression of GluN2B was changed without showing a notable difference in total GluN2B expression, raised the possibility that the ECM may regulate GluN2B dynamics. To test this possibility cortical neurons were stained with Anti-GluN2B antibody and the receptor’s dynamics were tracked over time. As anticipated, lack of ECM due to Hya treatment decreased GluN2B endocytosis, thus confirming the hypothesis that hyaluronic acid- based ECM regulates GluN2B dynamics through endocytosis.
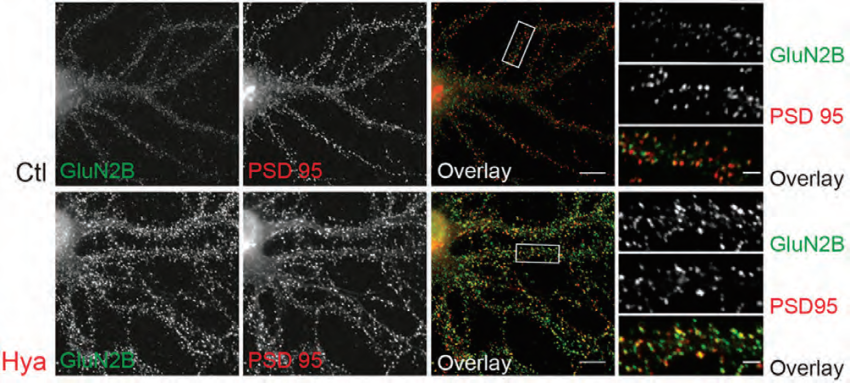
Immunocytochemistry of living rat dissociated hippocampal neurons. Extracellular staining of cell with Anti-NMDAR2B (GluN2B) (extracellular) Antibody (#AGC-003). GluN2B cell surface expression (green) increases following extracellular matrix (ECM) removal (lower panels). GluN2B expression coincides with PSD-95 synaptic marker.
Adapted from reference 17. with permission of SPRINGER NATURE.
The prefrontal cortex (PFC) is a special cortical region that regulates many behavioral skills including the ability of decision making and moderating impulsive behaviors9. This brain region often matures later than other cortical areas and therefore, many impulsive and risky behaviors that are associated in adolescence, are thought to be attributed to this characteristic. Differences in GluN2A protein levels between premature and mature PFC of young and adult mice were observed in western blot analysis using the respective Alomone Labs antibody; no notable difference in GluN2B was observed regardless of age and brain region (Figure 5)9. Hence, the authors conclude that this characteristic could partially explain the impulsive behavior in adolescence9.
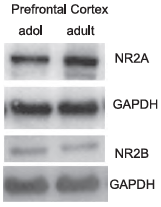
Western blot analysis of mouse brain prefrontal cortex (PFC) lysates using Anti- NMDAR2A (GluN2A) (extracellular) Antibody (#AGC-002) and Anti-NMDAR2B (GluN2B) (extracellular) Antibody (#AGC-003). GluN2A expression (upper panel) appears to increase with age, while that of GluN2B (lower panel) does not change. Adapted from reference 9 with permission of The American Physiological Society.
Noonan syndrome is a genetic disorder associated with developmental and learning disabilities14. A recent study examined how a point mutation in SHP protein (associated with Noonan syndrome) may contribute to cognitive dysfunction. In early developed cultured hippocampal neurons increased MAPK signaling due to SHP mutation increased the number and size of NMDAR receptors as evident by immunocytochemical staining of rat premature hippocampal dissociated neurons using Anti-NMDAR1 (GluN1) (extracellular) Antibody14. In midstage neurons (div12), MAPK signaling favored the expression of GluA1 as evident by immunostaining of cultured hippocampal neurons using Anti-GluR1 (GluA1) (extracellular) Antibody (#AGC-004)14. In mature neurons (div18) SHP mutations increased both the size and number of GluA1 receptors14. Overall these data suggest that MAPK signaling caused by SHP mutations can alter neuronal development and cause cognitive impairments.
NMDAR Expression in Brain Disorders
Ischemic brain injury can inflict serious brain damage which results in various types of disabilities due neuronal cell death13. Despite the large interface and support of glial cells to neuronal function and synaptic plasticity, it is now clear that glial cells are involved in several pathologies related to brain ischemia5, 13. Glial cells also express NMDARs, however, remodeling processes related to these ion channels are not fully understood5. NMDAR remodeling in response to ischemic brain injury in glial cells was investigated in part by immunohistochemical staining of mouse brain sections using Anti-NMDAR2C (GRIN2C) (extracellular) Antibody (#AGC-018) and Anti- NMDAR2D (GRIN2D) (extracellular) Antibody (#AGC-020). Staining showed that ischemic injury induced their expression in cortical brain slices5. Next, the authors tested whether this expression pattern can influence calcium transitions in cortical glial cells. Indeed, they noticed a persistent elevation in calcium concentration in response to NMDA receptor agonist application. Blocking sodium NaV channels with Tetrodotoxin (#T-500 or #T-550) did not influence calcium fluxes compared to specific NMDAR blockers, suggesting a causative link between NMDAR expression and function5.
Epilepsy and its related diseases such as epileptic aphasia have been associated with NMDAR dysfunction and GluN2A mutations18. In a paper by Sibarov et al. the relationship between several point mutations in GluN2A protein and surface expression was explored by means of immunofluorescent microscopy in HEK-293T cells18. Mutated GuN2A variants were coupled to mCherry reporter and co-expressed with GluN1-GFP. To quantify surface expression of wild-type GluN2A, HEK-293T cells were stained using Anti-GluN2A antibody from Alomone Labs under non-permeabilizing conditions and quantified the overlap between mCherry (mutant) and antibody (WT) staining18. Mutants with low cell surface expression displayed similar low electrophysiological recordings18.
Alzheimer’s disease is an age-related brain-disorder where amyloid beta aggregates are associated with cognitive decline and neurotoxicity. In Chinese medicine, Rhynchophylline (RIN) an herbal bioactive compound, is thought to have neuroprotective properties, but the mechanism remains elusive20. Using Anti-NMDA Receptor 2B antibody the authors found that RIN decreased extra-synaptic GluN2B expression when Dentate Gyrus (DG) neurons were exposed to amyloid beta protein20. Thus, given that extra- synaptic NMDARs mediate cytotoxic calcium cell-singling, inhibition of this pathway may partially explain RIN’s neuro-protective effects.
References
- Aloisi, E. et al. (2017) Nat. Commun. 8, 1103.
- Antonelli, R. et al. (2016) J. Neurosci. 36, 5437.
- Atkin, G. et al. (2015) J. Neurosci. 35, 6165.
- Dupuis, J.P. et al. (2014) EMBO J. 33, 842.
- Dzamba, D. et al. (2015) Cell. Mol. Neurobiol. 35, 1187.
- Guo, H. et al. (2017) Sci. Rep. 7, 11608.
- Jézéquel, J. et al. (2017) Nat. Commun. 8, 1791.
- Jézéquel, J. et al. (2017) Biol. Psychiatry 82, 766.
- Konstantoudaki, X. et al. (2018) J. Neurophysiol. 119, 822.
- Lau, C.G. and Zukin, R.S. (2007) Nat. Rev. Neurosci. 8, 413.
- Lesept, F. et al. (2016) Cell Death Dis. 7, e2466.
- Mikasova, L. et al. (2017) Sci. Rep. 7, 8053.
- Nedergaard, M. and Dirnagl, U. (2005) Glia 50, 281.
- Oh, J.Y. et al. (2017) Neurosci. Lett. 649, 41.
- Paoletti, P. et al. (2013) Nat. Rev. Neurosci. 14, 383.
- Piguel, N.H. et al. (2014) Cell Rep. 9, 712.
- Schweitzer, B. et al. (2017) Sci Rep. 7, 10991.
- Sibarov, D.A. et al. (2017) Front. Cell Neurosci. 11, 155.
- Wenthold, R.J. et al. (2003) Annu. Rev. Pharmacol. Toxicol. 43, 335.
- Yang, Y. et al. (2018) Neuropharmacology 135, 100.
- Zhu, S. et al. (2016) Cell 165, 704.