SCN5A is the gene encoding NaV1.5 voltage-gated Na+ channels. These membrane proteins are responsible for the initiation and propagation of the excitable cell action potential in the heart. They have been extensively studied over the years due to their critical importance in regulation of cardiac pulsing, and are the prime target for several important antiarrhythmic drugs. This article briefly reviews the contribution of Alomone Labs products to the understanding of NaV1.5 role and function at the cellular and physiological levels.
Introduction
Voltage-gated Na+ (NaV) channels are heteromultimeric, transmembrane proteins. They are abundant in the vertebrate heart, skeletal muscle and nerve tissues, where they enable the initiation and propagation of action potentials in excitable cells. They do so by allowing transmission of Na+ ions in response to membrane voltage alterations, thus executing essential processes such as neuronal firing and muscular contraction, which in turn permit higher events, e.g. locomotion and cognition, to occur9,18.
A plethora of pathologies (channelopathies), usually inherited, are attributed to defects in one or more mammalian NaV isoforms; from generalized epilepsy and familial infantile-seizures, through hyperkalemic periodic paralysis and variants of myotonia to congenital long QT syndrome, arrhythmia and even sudden infant death syndrome. Accumulating evidence is also being gathered regarding the crucial role Na+ channels play in cancer invasion12, and recently NaVs have even been identified as the primary mediators of abnormal gastrointestinal motility9,18,19.
NaV1.5 is expressed almost exclusively in cardiac tissues. It generates the rapid action potential (AP) of myocardia, and is responsible for its conduction and duration and contributes to the plateau of the AP through a continuous inward Na+ current (late INa; INaL)6,33. The channel alternates between three modes of conformation: activation, upon a rapid upward shift in membrane potential from the resting state, rendering the channel in the open conformation, followed by a stable, inactivation phase less than a millisecond later, and a closed conformation, at a resting membrane potential of about -85 mV. Conductance modifications and channel trafficking ascribed to genetic mutations of NaV1.5 and its regulatory partners are at the base of several inborn cardiac channelopathies1,3. The reviewed studies presented below aim to shed light upon some of the more severe mutant phenotypes, malfunctions and mechanisms of NaV1.5.
Heart Malfunctions
Brugada Syndrome/Decreased Na+ Density Mutants
Brugada syndrome (BrS), a rare clinical cardiac disorder characterized by an abnormal rise of the ST-segment in an ECG scan, usually unmasked by Na+ channel blockers20, is an arrhythmical disorder with episodes of ventricular fibrillation, which significantly elevates the risk of sudden cardiac death and sudden infant death syndrome14,15,20, albeit without structural heart disease. Although more than 90 mutations leading to BrS have been mapped to NaV1.5, the majority of which reduce INa either by alternations of its membrane expression density or its biophysical properties, only about 20% of the patients carry a mutation in the gene encoding it. In an effort to map causative factors of this elusive morbidity, Petitprez et al.20 investigated a novel BrS mutation, C1850S, which they identified by a molecular screening of SCN5A in a patient suffering from phenotypical BrS. Following a transfection of HEK293 cells with WT and mutant DNA, results of immunoblots using Anti-NaV1.5 (SCN5A) (493-511) Antibody (#ASC-005) showed similar levels of protein expression between WT and mutant strands (Figure 1), confirming the hypothesis that the mutation does not alter the channel’s membrane density, but rather, as was evident by electrophysiological experiments, reduces the peak of INa, its intermediate-density and the time it takes to inactivate20.
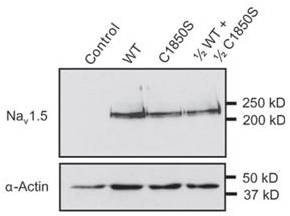
Western blot analysis using Anti-NaV1.5 (SCN5A) (493-511) Antibody (#ASC-005) of HEK293 cells expressing Wild Type and mutant NaV1.5 channel shows that the mutation does not affect the expression of NaV1.5.
Adapted from reference 20 with permission of Oxford Journals.
Using a different approach, Kattygnarath et al.14 studied NaV1.5’s regulatory associates, and focused on MOG1, a small (28 kDa) ubiquitous nuclear protein. MOG1 colocalizes with NaV1.5 at ventricular myocytes and interacts with it through the latter’s intracellular loop between domains II and III, and increases the Na+current NaV1.5 conveys. E83D, a MOG1 mutant, was transfected into adult rat cardiomyocytes in parallel with its WT counterpart. Immunofluorescence with Anti-NaV1.5 (SCN5A) (493-511) Antibody revealed that in E83D-transfected cells, NaV1.5 accumulates around the nucleus, with a significantly lower density in the cell’s membrane, even though the MOG1 mutant itself was distributed evenly along these locales. This supports their previous electrophysiological attempts in which the INa density was reduced in mutant cells14.
Investigating factors involved in NaV1.5 malfunction, Liu et al. hypothesized that NADH is a causative player in the downregulation of NaV1.5, by the observation that a mutation in GPD1-L (involved in NAD-dependent energy metabolism) induces BrS. Treating cells with Chelerythrine chloride (#C‑400), a known blocker of Protein Kinase C (PKC), they demonstrated that the INa reduction happens due to an increase in NADH concentration, since chelerythrine inhibited the decrease in NaV1.5 currents via PKC. Using Anti-NaV1.5 (SCN5A) (493-511) Antibody in fluorescent imaging, no significant differences were observed in the NaV1.5 membrane density and localization between cells incubated with agents known to increase NADH (thus reduce INa) and control samples15.
The D1275N SCN5A mutant is associated with several cardiac arrhythmic disorders. However, heterologous expression of this mutant does not display any functional anomalies, nor does it show altered expression levels. When expressed in a mouse model, the same mutant is expressed to lower levels and displays a decrease in Na+ currents in cardiomyocytes as shown by both western blot analysis and immunostaining with Anti-NaV1.5 (SCN5A) (493-511) Antibody. Overall, this study emphasizes the importance of the channel’s environment in modulating its activity28.
Prolonged Late-Na+ Current Complexities
A related abnormality is the long QT syndrome (LQTS). LQTS, a gain-offunction arrhythmia, is characterized by a lengthened action potential density (APD), a longer QT interval, and susceptibility to polymorphic v ntricular tachyarrhythmia (torsade de pointes; TdP)29. In this case, NaV1.5 fails to inactivate normally and maintains an elongated action potential plateau due to a prolonged INaL. Such an increase leads to a disruption of the cellular ionic homeostasis and results in an intracellular Ca2+ overload2,16,21. Interestingly, a gender-dependent bias is seen in the prevalence of LQTS and BrS: females, who seem to have a lower density of the cardiac K+ channel α subunit which supplies the current involved in atrial repolarization (IKr) as well as in its inactivation (ITo)25, are more prone to LQTS than males, who instead have a higher risk of BrS. However, the distinct arrhythmia profiles cannot be relied solely on the gender-specific differences in K+ currents. Speculating that a non-homogenous expression of NaV1.5 is the determining gender-related risk factor, a study was set out to explore the sex-biased distribution of NaVs; immunoblots using Anti-Pan NaV Antibody (#ASC-003) revealed an overall even NaV dispersion between the genders and thus led the authors to base the anomaly on the male hormone, testosterone, which they claimed to be a depressant of the INa amplitude4. However, Lowe et al. ruled out any interference of hormones, to show that NaVs are expressed evenly among the genders by using Anti-NaV1.5 (SCN5A) Antibody in immunoblots. ATX-II alone, a potent Na+ channel opener, drastically increased the INaL in females, whereas only moderately did so in males16. Their finding enforced a previous one, which also used ATX-II (#STA-700) to induce LQTS-like symptoms; this proves a direct relationship between increased INaL and LQTS27.
ΔKPQ is a SCN5A mutant which induces a long QT-like phenotype. Different inactivation inhibitors were tested on the mutant and was compared to its wildtype counterpart. Alomone Labs ATX-II was used and surprisingly inactivated the mutant channel by shifting the steady-state inactivation, as opposed to its activating effect on the wild type channel23.
Although lengthened QT interval often reflects TdP, each cannot reliably indicate the presence of the other. Hondeghem et al. used Alomone Labs ATX-II, along with dofetilide (Ikr blocker) to show that in isolated Langendorff perfused hearts, TdP occurs with prolonged, normal and even shortened APDs11.
Connexins and NaV1.5 are two important factors for impulse propagation in cardiac cells. As these parameters are often affected in cardiac disease, a study was initiated to see how these two factors can together affect conductivity. Heterozygous animals for NaV1.5 and/or connexin-43 (Cx43) were used; thereby leading to a decrease of up to 50% in protein expression (Anti-NaV1.5 (SCN5A) (493-511) Antibody was used in immunohistochemical studies to monitor NaV1.5 protein levels). The electrophysiological properties of these mutants showed that the deficiency of both conductance and impulse propagation, while caused a reduced Na+ current, surprisingly benefited the QT interval24. In a related study, elevated levels of Cx43 and NaV1.5 (shown using Anti-NaV1.5 (SCN5A) (493-511) Antibody) in the left ventricle (LV) were spotted in an experiment studying the effects of rabbit’s heart biventricular pacing (BIV). Increases in KVLQ 1 (a gene coding a cardiac KV7.1 K+ channel subtype) protein levels, observed using Anti-KCNQ1 Antibody (#APC-022), explained the shortened QT intervals occasionally seen in artificially paced hearts22.
Although NaV1.5 is the major voltage-gated Na+ channel isoform expressed in the heart, designated neuronal type NaV channels, namely NaV1.1, NaV1.3 and NaV1.6 have also been detected in the mammalian heart and contribute 10-20% of the Na+ current peaks in cardiomyocytes and Purkinje cells31. In a study, their contribution to heart failure was investigated. Using a combination of electrophysiological measurements and immunological staining using Alomone Labs Anti-SCN1A (NaV1.1) Antibody (#ASC-001), Anti-SCN3A (NaV1.3) Antibody (#ASC- 004), Anti-NaV1.5 (SCN5A) (493-511) Antibody (#ASC-005) and Anti-NaV1.6 (SCN8A) Antibody (#ASC-009), the results indicate that most NaV isoforms are expressed in cardiac ventricular myocytes (Figure 2), and that upon pressure overload induction (i.e. heart failure), there is an increase in TTX-sensitive currents, namely those of NaV1.1, NaV1.3 and NaV1.6 channels. This is reflected by an increase in the expression of NaV1.1 and NaV1.6 and a decrease of NaV1.5 channels31.
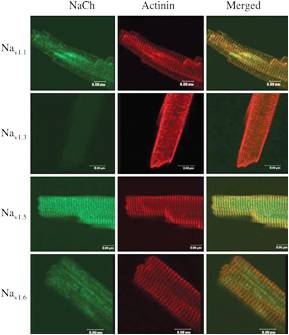
Immunocytochemical staining of rat cardiac ventricular myocytes using Anti-SCN1A (NaV1.1) Antibody (#ASC-001), Anti-SCN3A (NaV1.3) Antibody (#ASC-004), Anti-NaV1.5 (SCN5A) (493-511) Antibody (#ASC-005) and Anti-NaV1.6 (SCN8A) Antibody (#ASC-009).
Adapted from reference 31 with permission of Oxford Journals.
Drugs acting on NaV1.5
Some antiarrythmic agents, while not altering the QT interval, have nonetheless a critical impact on TdP risk. One such agent, amiodarone, a drug which elevates the risk to TdP in patients with the S1102Y SCN5A polymorphism (which heightenes INaL peak and duration), was reported to facilitate an already prolonged APD in rabbits all the while reducing its peak (by inhibiting the K+ current Ikr rather than that of NaV) when acutely administrated at low doses; an alarming finding which called for an immediate reassessment of the drug’s safety. The S1102Y SCN5A polymorphism was mimicked by administering Alomone Labs ATX-II30. A related study, focusing on mexilethine, a NaV blocker, showed that in some of the SCN5A gain-of-function mutants, treatment with mexiletine can worsen the symptoms of a LQTS-variant by causing an increased trafficking of NaVs to the membrane – as was observed using Anti-NaV1.5 (SCN5A) (493-511) Antibody in immunostaining21.
Bepridil has multi-channel blocking properties and exhibits anti-electrical remodeling effects in the diseased heart. A study aimed at investigating the long term effects of bepridil in rat cardiomyocytes and heterologous human NaV1.5 system, found that bepridil inhibited INa in the short term, and stimulated the channel in the long run. NaV1.5 channel levels were monitored using Anti-NaV1.5 (SCN5A) (493-511) Antibody (#ASC-005) and Anti-SCN2B (NaVβ2) Antibody (#ASC-007). The long term enhancing effect was seemingly achieved by inhibiting the proteasome activity in a Ca2+/calmodulin (Ca2+/CaM) dependent signaling pathway; western blot analysis showed an increase in NaV1.5 (but without an upregulation of its beta subunits) upon bepridil treatment, attributed to the proteasome-inhibiting effect of the drug. The authors concluded that while bepridil-induced cardio-aversion does occur in the long term in diseases related to atrial remodeling, it has an unpredicted pharmacological outcome due to its intereference with the Ca2+/CaM pathway13. F15845, a newly discovered NaV1.5 channel blocker, was shown to prevent hypoxia-induced contraction of the femoral artery. The Na+ channel is prominently expressed in the femoral artery, as shown using Anti-NaV1.5 (SCN5A) (493-511) Antibody in immunofluorescent assays5.
Kinases in Cardiac Pathologies
Two recent studies have elucidated the role of calmodulin-dependent protein kinase II (CaMKII) regarding INaL. In heart failure, CaMKII is up-regulated and maintains a partly positive-feedback relationship with NaV1.5 such that INa promotes CaMKII auto-phosphorylation via Ca2+, which results in either a prolonged INaL, or a reduced INa at low and high heart rates, respectively. INAL induction via ATX-II treatment led to CaMKII activation which could be reversed by specific siRNA downregulation of NaV1.5 and not by NaV1.1 and NaV1.2 downregulation (Figure 3)2,32.

The decrease in NaV1.5 protein levels by siRNA reduces ATX-II-induced phosphorylation of CaMKII. NaV1.5 levels were monitored by using Anti-NaV1.5 (SCN5A) (493-511) Antibody (#ASC-005). Alomone Labs ATX-II (#STA-700) was used to activate NaV1.5 channels.
Adapted from reference 32 with permission of the American Physiological Society.
Serum-and glucocorticoid-regulated kinase-1 (SGK1), a PI3-kinase member is activated in prolonged APD arrhythmia. Immunoprecipitation experiments validated the binding of SGK1 to NaV1.5 (using in part Anti-NaV1.5 (SCN5A) (493-511) Antibody). Whole-cell patch clamp protocols of cardiomycytes expressing constitutively active SGK mutant displayed a 3.6 fold increase in INaL, compared to wild-type cells8.
The Effects of Reactive Oxygen Species and NCX
Reactive oxygen species (ROS) leads to an increase in INaL. The resulting increase in intracellular Na+ reverses the activity of Ca2+/Na+ exchanger (NCX) thus leading to Ca2+ overload (which results in long-QT and related arrhythmia). In isolated rat heart, administration of Alomone Labs ATX-II had the overall same effect as that of H2O2; both caused an increase in ROS, an increase in INaL, and subsequently a Ca2+overload. However, the mechanisms used are different26. A more recent study investigated the effects of ROS on the transcription regulation of NaV1.5. It seems that, upon oxidative stress, Foxo1 transcription factor binds to the promoter region of SCN5A and suppresses its expression. This was apparent in western blot analysis using Anti-NaV1.5 (SCN5A) (493-511) Antibody; NaV1.5 levels were significantly reduced in the presence of Foxo1, and high when Foxo1 RNA was silenced17. NCX plays an important role in excitation-contraction coupling in the neonatal rabbit heart, where Na+ influx is large enough to promote Ca2+ entry via NCX. This event is possible due to the co-localization between various NaV channels, including NaV1.5 (detected with Anti-NaV1.5 (SCN5A) (1978-2016) Antibody (#ASC-013)) in neonatal rabbit hearts. Reverse-mode activity of NCX is lost during adulthood because the spatial organization of NaV channels with respect to NCX changes10.
NaV1.5 in Cancer
Accumulating evidence suggests that NaVs have a pivotal role in the invasiveness of several types of cancer. A study mapped the functionality and expression of NaV1.5 in colon cancer and showed, in immunohistochemical staining using Anti-NaV1.5 (SCN5A) (493-511) Antibody, the abundance of the channel in the luminal surface of human malignant colon cells, and its absence in normal analogs12.
Because of its dependence on the cytoskeletal framework, Na+ channels can be impaired when treated with anti-cancer drugs like taxol (TXL), which polymerizes tubulin, an essential cytoskeletal protein. Indeed, patients treated with the drug often develop cardiac disorders. Casini et al. nicely demonstrated electrophysiologically the reduction of INa peak and density in cardiomyocytes treated with TXL and assured this observation was due to the channel’s reduced expression, in immunostaining procedures (Figure 4). They also established the necessity of the β1 subunit in enhancing INa7.

Immunocytochemical staining of neonatal rat cardiomyocytes (NRCs) using Anti-NaV1.5 (SCN5A) (493-511) Antibody (#ASC-005). Untreated cardiomyocytes (left) show NaV1.5 staining (green) in the cytoplasm and the membrane surface. Following taxol treatment, less NaV1.5 staining is observed and the channel is mostly localized to the cytoplasm.
Adapted from reference 7 with permission of Oxford Journals.
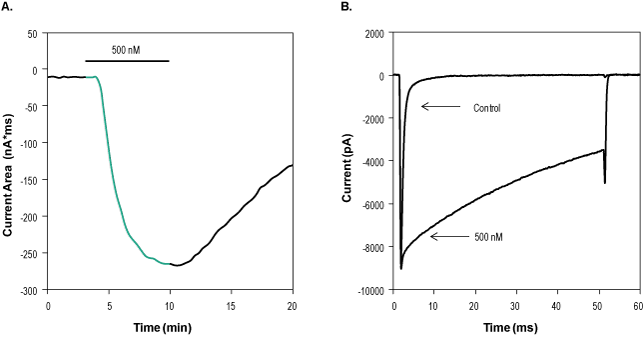
A. Time course of Jingzhaotoxin-II (#STJ-150) action. Current area was plotted as a function of time. Holding potential was -100 mV and currents were stimulated every 20 seconds by a voltage step of 50 msec from holding potential to -20 mV. 500 nM Jingzhaotoxin-II was perfused in the period marked by the horizontal bar (green), indicating a toxin-dependent decrease in NaV1.5 currents inactivation. B. Superimposed traces of NaV1.5 currents before and during 7 min application of 500 nM Jingzhaotoxin-II.
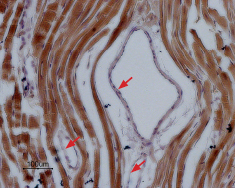
Immunohistochemical staining of NaV1.5 in rat myocardium paraffin-embedded section using Anti-NaV1.5 (SCN5A) (1978-2016) Antibody (#ASC-013), (1:100). Staining is specific for cardiomyocytes while smooth muscles cells in the artery walls are negative (red arrows). Hematoxilin is used as the counterstain.
References
- Abriel, H. (2007) Cardiovasc. Res. 76, 381.
- Ashpole, N.M. et al. (2012) J. Biol. Chem. 287, 19856.
- Balser, J.R. (1999) Cardiovasc. Res. 42, 327.
- Barajas-Martinez, H. et al. (2009) Cardiovasc. Res. 81, 82.
- Bocquet, A. et al. (2010) Br. J. Pharmacol. 161, 405.
- Carmeliet, E. (1987) Pflugers Arch. 408, 18.
- Casini, S. et al. (2010) Cardiovasc. Res. 85, 691.
- Das, S. et al. (2012) Circulation 126, 2208.
- George, A.L. (2005) J. Clin. Invest. 115, 1990.
- Gershome, C. et al. (2011) Am. J. Physiol. 300, H300.
- Hondeghem, L.M. et al. (2010) Naunyn Schmiedebergs Arch. Pharmacol. 382, 367.
- House, C.D. et al. (2010) Cancer Res. 70, 6957.
- Kang, L. et al. (2009) Br. J. Pharmacol. 157, 404.
- Kattygnarath, D. et al. (2011) Circ. Cardiovasc. Genet. 4, 261.
- Liu, M. et al. (2009) Circ. Res. 105, 737.
- Lowe, J.S. et al. (2012) Cardiovasc. Res. 95, 300.
- Mao, W. et al. (2012) PLoS One 7, e32738.
- Marban, E. et al. (1998) J. Physiol. 508, 647.
- Mazzone, A. et al. (2008) J. Biol. Chem. 283, 16537.
- Petitprez, S. et al. (2008) Cardiovasc. Res. 78, 494.
- Ruan, Y. et al. (2010) Circ. Res. 106, 1374.
- Saba, S. et al. (2010) Circ. Arrhythm. Electrophysiol. 3, 79.
- Spencer, C.I. (2009) Toxicon 53, 78.
- Stein, M. et al. (2009) Cardiovasc. Res. 83, 52.
- Van Wagoner, D.R. et al. (1997) Circ. Res. 80, 772.
- Wang, L. et al. (2008) J. Mol. Cell Cardiol. 45, 787.
- Wasserstrom, J.A. et al. (2009) J. Pharmacol. Exp. Ther. 331, 382.
- Watanabe, H. et al. (2011) Circulation 124, 1001.
- Wenzel-Seifert, K. et al. (2011) Dtsch. Arztebl. Int. 108, 687.
- Wu, L. et al. (2008) Cardiovasc. Res. 77, 481.
- Xi, Y. et al. (2009) Eur. J. Heart Fail. 11, 749.
- Yao, L. et al. (2011) Am. J. Physiol. 301, C577.
- Zygmunt, A.C. et al. (2001) Am. J. Physiol. 281, H689.