Epithelial cells form a dynamic barrier that separates different physiological compartments. On one hand they provide a physical barrier that prevents pathological microbes from entering the host and on the other hand they must maintain tissue homeostasis by allowing the regulated transport of solutes, electrolytes and water across them. In the present review we will focus on the main ion and water channels responsible for the normal epithelial function in three main organs: the colon, the lung and the kidney.
Introduction
Epithelial cells from different tissues perform specialized functions such as salt and water absorption in the kidney, mucus secretion in the colon and fluid reabsorption in the lung shortly before birth. Despite the diverse array of specific functions that epithelial cells perform in different tissues, there is a remarkably consistency in the mechanisms used by the cells. First of all, epithelial cells must be polarized in order to function properly. This is achieved by the differential distribution of membrane proteins along the apico-basal axis. The apical surface faces the lumen or external environment while the basolateral membrane faces the internal compartments. The net transport of solutes, ions and water across the epithelial membrane is then achieved by the coordinated action of transporters, ion channels and water channels unevenly distributed in the apical or basolateral membrane. The functional significance of these channels for the optimal epithelia tissue function is highlighted by the continuing realization that mutations in these molecules are the underlying cause of several human diseases (See Table).
Ion Channels in Epithelial Tissue
One of the main functions of epithelial tissue in the kidney and colon is to absorb about 90% of the salt and water entering the body. This is needed in order to maintain constant extracellular fluid volume and adequate blood circulation and hence blood pressure. Electrolyte regulation is also essential for the maintenance of airway surface fluid that facilitates optimal gas exchange in the lungs. In addition, a tight regulation of Na+ and K+ ions is needed to maintain optimal metabolism. For example, a mere 1-2% movement of K+ from the intracellular to the extracellular compartment can be potentially fatal.
The regulated movement of ions and water across epithelial membranes is accomplished by the coordinate function of a number of ion channels, transporters and pumps in the epithelial cells. In the last few years there is an enhanced understanding of the molecular identity of the transporter elements that function in epithelial tissue. In this review we will focus on the identity and function of the ion and water channels in epithelial tissue, and although the concerted function of several pumps and co-transporters is equally important, they won’t be treated here. Figure 2 shows the main ion channels present in colon, lung and kidney. Ubiquitously expressed ion channels such as CLC2 and CLC3 or the water channels AQP1 and AQP3 have been omitted for clarity.
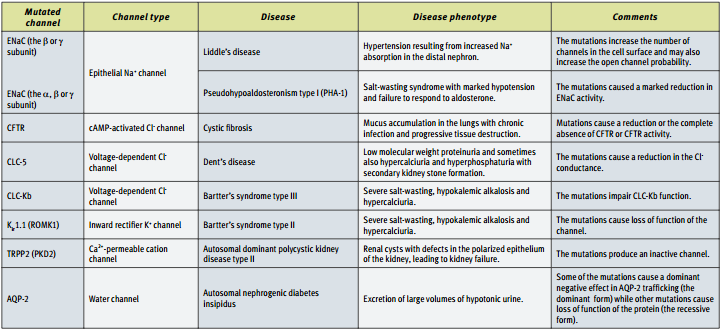
Mutations in Epithelial Ion Channels that Cause Human Diseases.
Na+ Channels in Epithelial Tissue
It is generally understood that the key determinant of epithelial water and salt absorption is the rate of Na+absorption. The greater part of the NaCl absorbed by the epithelium is electroneutral, mainly via the Na+/H+exchanger (NHE) and the Cl–/HCO3– exchanger that are situated at the apical (or luminal) side of the epithelium membrane. These two exchangers will not be discussed in this review. Na+ is also absorbed by electrogenic means via ion channels situated in the apical membrane, along a very favorable electrochemical gradient. The absorbed Na+ is then pumped out through the Na+-K+-ATPase located at the basolateral membrane, thus creating the driving force for additional Na+ uptake. The favorable Na+ electrochemical gradient is also maintained by the coordinated function of several transporter and ion channels, notably basolateral K+ channels and apical Cl– channels1.
ENaC
Electrogenic absorption of Na+ in the colon, kidney and lung is mediated by the amiloride-sensitive epithelial Na+ channel (ENaC). The ENaC channel consists of three different subunits that are all members of the ENaC family: the α, β and γ subunits. A fourth member of the family, the δ subunit, has been identified in humans (although not in rat and mouse). It can substitute for the α subunit to form functional channels with the β and γ subunits and it is mainly expressed in the brain2. The functional ENaC channel in epithelial tissue is therefore a heteromer with a presumed stoichiometry of α2βγ3. The α subunit is capable of producing small Na+ currents when expressed alone in Xenopus oocytes, whereas expression of the β or γ subunits alone produces no current at all. Co-expression of either β or γ with the α subunit increases the current by three to five fold while expression of all three subunits produces a current increase of a hundred fold3. Thus, optimal ENaC function depends on the correct assembly of all subunits. The ENaC subunits have a conserved topology consisting of two membrane-spanning domains with intracellular N and C-termini and a large glycosylated extracellular region4. The functional ENaC channel has very distinctive biophysical characteristics: the channel is voltage independent with very long closing and opening times. The ENaC channel is highly selective for Na+ over K+and as it name implies, is blocked by the diuretic compound amiloride (and its analogs)5,6.
The central role of ENaC in the regulation of Na+ homeostasis and hence blood pressure is underscored by the identification of two human diseases that arise from either gain- or loss-of-function mutations of the ENaC channel (See Table). Liddle’s syndrome is an inherited form of hypertension that stems from a dominant mutation of the ENaC channel (in either the β or γ subunits) that results in excessive activity of the channel and hence increased Na+ absorption7,8. Conversely, pseudoaldosteronism type I (PHA) is a dysfunction characterized by hypotension due to poor Na+ absorption that is associated with loss-of-function mutations which may occur in each of the three ENaC subunits9. These diseases are indicative of the importance of maintaining systemic Na+ homeostasis and thus, ENaC function, under tight control. This is achieved by the close regulation of ENaC expression and function through several mechanisms that are becoming more evident.
The main mechanism that regulates ENaC function is the control of the channel expression at the cell surface, since the open probability of the channel is very high (see figure 1). This is achieved through the binding of surface ENaC channels to Nedd4-2, a protein that is a member of the E3 ubiquitin protein-ligase family. Ubiquitination of ENaC tags the protein for internalization and degradation, therefore making Nedd4-2 a negative regulator of ENaC function10. Indeed, the mutations causing Liddle’s syndrome are mutations in the ENaC region responsible for binding to Nedd4-2: the PY motifs located at the intracellular C-termini of the β and γ subunits11. Thus, in Liddle’s syndrome, the mutated channels fail to associate to Nedd4-2 and undergo degradation resulting in enhanced expression of surface ENaC channel. Conversely mutations that cause PHA are mutations that interfere with the trafficking of ENaC to the cell surface resulting in decreased expression of the channel in the surface and therefore decreased Na+ absorption.
The main regulator of systemic Na+ homeostasis is the mineralocorticoid hormone aldosterone (ADH). Aldosterone regulates Na+ homeostasis through a classical feedback inhibition mechanism. High plasma Na+ levels induce the synthesis and release of aldosterone from the adrenal cortex. In aldosterone- responsive epithelia such as the distal colon and the kidney aldosterone enters the cell and binds with high affinity to the mineralocorticoid receptor (MR) and with low affinity to the glucocorticoid receptor (GR). The hormone-receptor complexes translocate into the nucleus and initiate a molecular cascade that results in the increase of apical membrane expression of ENaC and consequently increased Na+ absorption. Finally, the ensuing low levels of Na+ will then reduce the levels of aldosterone and close the metabolic feedback5,6. The precise molecular mechanisms that mediate aldosterone-induced enhanced ENaC function have been the object of intense study. One of the mechanisms by which aldosterone induces increased ENaC expression is by increasing transcription of the channel subunits and hence protein synthesis. However, the increase in Na+ absorption in response to aldosterone occurs before any significant rise in ENaC transcription can be detected, suggesting that aldosterone may induce the transcription of other proteins that regulate ENaC trafficking. One such protein is the serine-threonine kinase SGK1. SGK1 modulates ENaC surface expression at least in part through its interaction with Nedd4-2. SGK1 binds and phosphorylates Nedd4-2 that, as a result, is unable to bind to EnaC and subsequently there is a reduction in ENaC ubiquitination and degradation12. ENaC activity and hence Na+absorption can be also modulated by other hormones such as vasopressin and insulin through mechanisms not well understood but that likely involve activation of the cAMP and PI3-K pathways12,13.
One of the mechanisms that control ENaC activity via cAMP involves the cystic fibrosis transmembrane regulator (CFTR ) channel that is co-expressed with the ENaC channel in the apical membranes of the airway and colonic epithelium. Activation of CFTR following an intracellular rise in cAMP reduces ENaC-mediated Na+ absorption. Conversely, cystic fibrosis patients that have reduced expression of the CFTR channel showed increased ENaC activity that has been linked to the pathophysiological symptoms of the disease14,15.
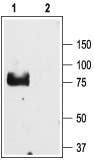
Western blotting of rat lung lysate using Anti-ENaC γ (SCNN1G) (extracellular) Antibody:
1. Anti-ENaC γ (SCNN1G) (extracellular) Antibody (#ASC-011) (1:200).
2. Anti-ENaC γ (SCNN1G) (extracellular) Antibody, preincubated with the negative control antigen.
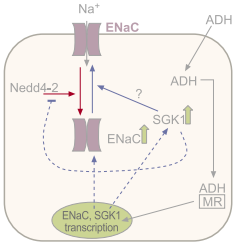
Figure 1. Regulation of ENaC Expression by Aldosterone (ADH). Schematic representation of ENaC regulation by aldosterone (ADH). ADH enters the cells and binds to the mineralocorticoid receptor (MR). The complex translocates into the nucleus where it upregulates ENaC transcription. In addition, in a much rapid response, there is increased surface expression of ENaC, due to the actions of the serine-threonine kinase SGK1 that phosphorylates and inactivates Nedd4-2 a protein that mediates ENaC internalization.
Cl– Channels in Epithelial Tissue
As the several human diseases caused by mutations in Cl– channels (See Table) can attest, these channels are involved in several crucial processes such as cell volume and membrane potential regulation, transepithelial transport and organelle acidification. In spite of their importance in epithelial function, Cl– channels in general are less understood than other ion channels. Here is a brief summary of the most salient epithelial Cl– channels. Cl– channels such as CLC-2 and CLC-3 that are ubiquitously expressed in epithelia but whose function is less understood, won’t be discussed here.
A. CFTR
The most dominant Cl– channel in several epithelial tissues, especially in lung and colon is the cystic fibrosis transmembrane regulator (CFTR). CFTR is a member of the ATP-binding cassette (ABC) transporter superfamily. This large family that comprises 48 known human members uses ATP hydrolization as the driving force for the translocation of a wide variety of substrates (including sugars, amino acids, proteins and hydrophobic compounds) across cellular membranes. The CFTR is unique among ABC transporters in that it is a cAMP-regulated Cl– channel16, 17. It shares the superfamily topology of 12 transmembrane domains with two nucleotide-binding domains (NBDs) and a regulatory (R) domain in the large third intracytoplasmic loop that is phosphorylated in multiple sites by PKA. Mutations in the CFTR gene cause channel dysfunction in several ways, ranging from complete loss of surface expression to diminished Cl– secretion. Defects in the CFTR gene cause cystic fibrosis, the most common genetic disease among Caucasians, as well as a form of male sterility.
Regulation of the CFTR channel is accomplished through the activation of surface receptors that couple to adenyl cyclase, raise cAMP cellular levels and thus activate PKA. This has been demonstrated for the β2 adrenergic and adenosine receptors and the vasopressin hormone among others18.
Besides enhanced Cl– conductance, activation of CFTR also leads to the regulation of other ion channels. The best-studied case is its interaction with the ENaC channel as mentioned above, although it can probably regulate other ion channels as well (see Kir1.1 below for example). The mechanism by which CFTR regulates other ion channels is not clear, but it may involve protein-protein interactions via molecules that interact with its C-terminal PDZ binding motif, such as the NHERF adaptor protein19.
B. CLC-Kb
The CLC-Kb channel is a member of the voltage-dependent Cl- channel superfamily that is almost exclusively expressed in renal epithelial cells20. CLC-Kb is localized at the basolateral membrane of the thick ascending loop of Henle, a nephron segment involved in NaCl reabsorption. In this kidney segment, NaCl is taken up apically by the combined activity of the Na+-K+-2Cl– co-transporter (NKCC2) and the Kir1.1 K+ channel. Cl– then exits the cell through the CLC-Kb channel. Mutations in these three genes have been implicated in Bartter’s syndrome (types I to III), highlighting again the importance of the coordinated action of ion channels and transporters for transepithelial ion transport21. CLC-Kb needs the β subunit barttin to function properly. Barttin is the only known β subunit for CLC channels and is probably involved in the regulation of surface expression of CLC-Kb. In addition, expression of barttin in combination with CLC-Kb causes increased currents that are enhanced by extracellular Ca2+ and inhibited by low extracellular pH22.
C. CLC-5
The CLC-5 channel is another member of the CLC voltage-dependent Cl– channel superfamily. CLC-5 is predominantly expressed in the kidney although it has been found also in intestine, brain and liver. Mutations in CLC-5 cause Dent’s disease, a disorder with quite variable symptoms that may include hypercalciuria and hyperphosphaturia and kidney stones. The most consistent symptom is low-molecular weight proteinuria and is the best understood in the context of loss of function mutations in the CLC-5 channel.
Renal reabsorption of proteins such as albumin is accomplished via a characteristic receptor-mediated endocytosis mechanism. Albumin is the ligand of the megalin-cubulin scavenger receptor. The albumin-receptor complex is internalized into coated clathrin pits that progress from early to late endosomes. At this stage, the intraendosomal fluid is acidified by the H+-ATPase and the albumin is degraded in the lysosome. CLC-5 was found to be located intracellularly, particularly in endosome vesicles in renal proximal tube cells, where it colocalizes with the H+-ATPase pump20. It appears that efficient endosomal acidification depends on intravesicular influx of Cl–, probably because it dissipates the transmembrane potential generated by the proton pump23. Indeed, in CLC-5 mouse knock out models defective endocytosis and vesicle acidification has been described, corresponding with the observed phenotype of human Dent’s disease24, 25.
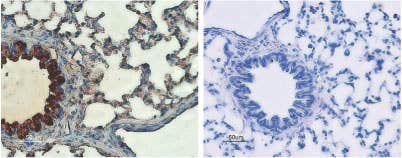
Expression of CFTR in rat lung. Rat lungs sections were incubated with Anti-CFTR Antibody (#ACL-006) (upper panel). Strong staining of bronchial epithelial cells appears red (black arrow) while lighter staining of alveolar cells appears red-brown (red arrow). There is also positive staining of macrophages while smooth muscle and endothelium are negative. Counterstain of cell nuclei appears blue. A negative control is shown in the lower panel.
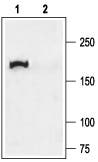
Western blotting of rat lung membranes:
1. Anti-CFTR Antibody (#ACL-006) (1:200).
2. Anti-CFTR Antibody, preincubated with the negative control antigen.
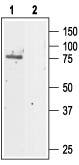
Western blotting of rat kidney lysate:
1. Anti-CLC-5 Antibody (#ACL-003) (1:200).
2. Anti-CLC-5 Antibody, preincubated with the negative control antigen.
K+ Channels in Epithelial Tissue
K+ channels situated at the basolateral membrane play an important role in epithelial cells by stabilizing the membrane potential and maintaining the driving force for the electrogenic transport of Na+ and Cl–. The luminal K+ channels are important for the excretion of K+ in secreted fluids, and are therefore involved in regulating K+ body homeostasis.
In some epithelial tissues, notably in the lung, the identity of the K+ channels involved in transepithelial transport is not clear. Electrophysiological and in some cases immunocytochemistry experiments have revealed the presence of several voltage-dependent K+ channels in alveolar epithelial cells such as KV1.1, KV1.3, KV1.4, KV4.2 and KV4.3 whereas conflicting results have been found regarding the expression of Ca2+-dependent K+ channels and ATP-sensitive K+ channels in these cells26, 35.The best characterized K+channels in epithelial tissue will be discussed below.
A. Kir1.1
Kir1.1 (ROMK1, KCNJ1) was the first member of the family of inward rectifying K+ channels to be cloned. The family members are characterized by a K+ efflux that is limited by depolarizing membrane potentials thus making them essential for controlling resting membrane potential and K+ homeostasis. The family’s topology consists of two transmembrane domains that flank a single and highly conserved pore region with intracellular N- and C-termini27. Kir1.1 is mostly expressed in the apical membrane of several kidney segments such as the thick assembly limb (TAL) and the cortical collecting duct (CCD). As mentioned above, loss of function mutations in the Kir1.1 gene cause Bartter’s syndrome type II, due to the impairment of K+ efflux and the subsequent inability of the NKCC2 transporter to continue NaCl uptake28. As the open probability of the Kir1.1 channel is very high, regulation of channel activity is largely achieved by controlling channel expression at the cell surface. The molecular mechanisms that control Kir1.1 trafficking are less well understood than the ones controlling the ENaC channel trafficking, although they show several similarities. Kir1.1 enhanced surface expression is dependent on cAMP-induced direct PKA phosphorylation, while src –dependent tyrosine phosphorylation is involved in Kir1.1 downregulation. Indeed, the vasopressin hormone that induces an increase in cAMP in selected epithelial renal cells is known to increase K+ efflux. Similarly, SGK1 a serine-threonine kinase regulated by aldosterone has been shown to directly phosphorylate Kir1.1 and hence increase surface channel expression29, 30.One of the most puzzling observations concerning the native renal apical K+ channel is it sensitivity to ATP. This is because the Kir1.1 channel shows no ATP sensitivity when expressed in heterologous systems, leading to the conclusion that the native channel may include an additional “regulatory” subunit. One candidate is the CFTR channel that may directly or indirectly (via NHERF-1 and NHERF-2 adaptor proteins, for example) bind Kir1.1 and confer it with ATP sensitivity31.
B. KV7.1
KV7.1 (KCNQ1) is a member of the voltage-dependent K+ channel superfamily. The channel was first identified as the underlying molecular defect causing sudden death from cardiac arrhythmia. The native cardiac channel was found to consist of KV7.1 together with the β subunit KCNE1 (Isk). In epithelial tissues, chiefly in the colon, KV7.1 was found to associate with KCNE3 (MIRP2) to form the small-conductance cAMP-sensitive K+ channel that had been previously identified by pharmacological data32. In the colon, the KV7.1/KCNE3 K+ channel is located at the basolateral membrane where it has a role in maintaining Cl– secretion by providing the necessary driving force for Cl– luminal exit33. Thus, a typical Cl– secretion inducer such as vasopressin binds to its receptor and causes a rise in intracellular cAMP that activates both the CFTR Cl– channel and the KV7.1/KCNE3 K+ channel. The activation of the latter channel will greatly facilitate Cl- exit through the CFTR channel. The KV7.1 channel together with KCNE3 or another β subunit, may also play a role in Cl– secretion in other epithelial tissues such as kidney and lung, although the molecular composition of the native channel in these tissues is not well defined.
C. KCa3.1
KCa3.1 (SK4) is a Ca2+-dependent K+ channel that like KV7.1 hyperpolarizes the cell membrane and thereby increases the driving force for electrogenic ion transport. KCa3.1 is a part of the Ca2+-dependent K+ channel subfamily that is totally voltage-independent and is activated by a rise in intracellular Ca2+. The channel has a similar topology to that of KV channels, that is six transmembrane domains and intracellular N- and C-termini. KCa3.1 is abundantly expressed in colonic epithelium and perhaps in other epithelial tissue as well34. The channel is largely responsible for maintaining the membrane resting potential, while Ca2+-elevating agonists such as acetylcholine will greatly increase the open probability of the channel and thus support electrogenic transport of Na+ or Cl-35.
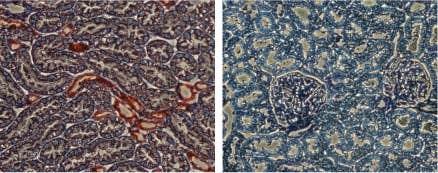
Expression of Kir1.1 in rat kidney. Rat kidney sections were incubated with Anti-KCNJ1 (Kir1.1) Antibody (#APC-001) (upper panel). There is strong staining (red) of tubular epithelial cells in distal tubes. Note that no staining is observed in proximal tubules (arrows). Counterstain of cell nuclei appears blue. A negative control is shown in the lower panel.
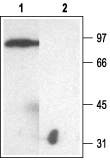
Western blotting of rat kidney membranes:
1. Anti-KCNJ1 (Kir1.1) Antibody (#APC-001) (1:200).
2. Anti-KCNJ1 (Kir1.1) Antibody, preincubated with the negative control antigen.
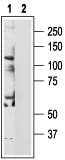
Western blotting of PC3 cell line lysate:
1. Anti-TRPV6 Antibody (#ACC-036) (1:200).
2. Anti-TRPV6 Antibody, preincubated with the negative control antigen.
Ca2+ Channels in Epithelial Tissue
Ca2+ serves as a second messenger in several crucial physiological functions such as neuronal excitability, muscle contraction, bone formation and programmed cell death. Thus, Ca2+ homeostasis is tightly regulated with Ca2+ levels maintained at near constant levels in the blood and extracellular fluid. A good example of how Ca2+ disregulation can affect normal organ function is found in the inherited human disorder polycystic kidney disease (PKD) that is caused by mutations in the PKD1 and PKD2 (now named TRPP1 and TRPP2 respectively) genes (See Table and below).
A. TRPV5 and TRPV6
All the Ca2+ in our bodies is derived from our diet and therefore the mechanisms of Ca2+ absorption must be highly regulated. Almost all dietary Ca2+ is absorbed in Ca2+ absorbing epithelia such as intestine and kidney. As a first step, Ca2+ enters the epithelial tissue via apical Ca2+ receptors. Since high levels of intracellular Ca2+can be toxic, the absorbed Ca2+ is buffered by the calbindin proteins and then extruded at the basolateral membrane by the Na+-Ca2+ exchanger (NCX1, in the kidney) or the Ca2+-ATPase pump (mostly in the intestine) by an energy-consuming process. It is only recently that the molecular nature of the apical Ca2+receptors has been established, with the cloning of TRPV5 (also known as ECaC1) and TRPV6 (or CaT1) from the kidney and intestine respectively36,37.
TRPV5 and TRPV6 are highly homologous (about 75% amino acid identity) members of the large TRP superfamily. They belong to the TRPV (vanilloid) subfamily named after the first identified member, the vanilloid receptor 1 (TRPV1). The channels display the signature topology of six transmembrane domains, a putative pore-forming region between transmembrane domains 5 and 6 and large intracellular N- and C- termini. In terms of expression it is generally assumed that TRPV5 is preferentially expressed in the kidney whereas TRPV6 is more prominently expressed in the intestine. However, expression of both channels has also been detected in placenta, prostate, pancreas, salivary gland and colon. The channels are both ligand and voltage independent with Ca2+ entering the cells along a very favorable electrochemical gradient. A feedback inhibition mechanism prevents intracellular Ca2+ from reaching toxic levels: an increase in intracellular Ca2+results in channel inactivation. Since TRPV5 and TRPV6 are the rate-limiting step for epithelial Ca2+ uptake their expression is regulated by the biologically active form of vitamin D1, α, 25-dihydroxy-vitamin D3, the key regulator controlling Ca2+ absorption. Increased levels of dietary Ca2+ induce an increase in 1α, 25- dihydroxy-vitamin D3 levels which in turn generate an increase in mRNA expression of both channels and hence, an increase in Ca2+ absorption38.
B. TRPP1 and TRPP2
As mentioned above, mutations in TRPP1 (also known as polycystin 1 or PKD1) and TRPP2 (polycystyn 2 or PKD2) are the underlying cause of the dominant form of polycystic kidney disease (PKD) a common condition (with a frequency of 1:500) that leads to progressive renal failure39. Although both proteins have been recently classified as members of the TRP superfamily (TRPP subfamily) only TRPP2 is thought to form a non-selective Ca2+-permeable channel whereas TRPP1 is thought to interact with TRPP2 and regulate its activity40. How mutations in both proteins can produce renal cysts that are the hallmark of PKD is not well understood. It has been observed that the balance between proliferation and apoptosis that is essential for normal organ function is disturbed in polycystic kidneys suggesting that a defect in TRPP2-mediated Ca2+ signaling may be involved. In addition, alterations in the polarity of renal epithelia has been detected with aberrant location of proteins such as Na+/K+-ATPase, EGF receptors, cathepsin B and matrix metalloproteinase 2 in the apical rather than the basolateral membrane39. The subcellular location of both TRPP1 and TRPP2 is not completely resolved with reports showing both apical and intracellular expression. The current model, though, proposes that TRPP1 functions as a membrane receptor that can bind to different proteins, carbohydrates and lipids and act as a mechanosensor that transduces different signals with the help of TRPP2-mediated Ca2+ signaling41.
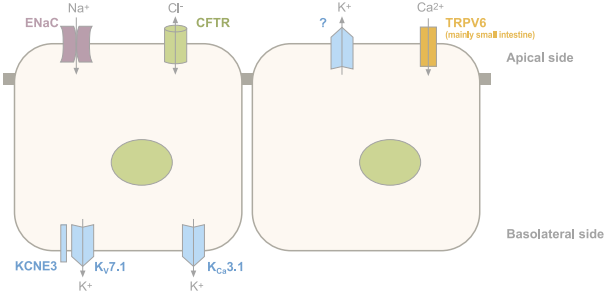
Figure 1a: Ion channels in colon epithelial cells.
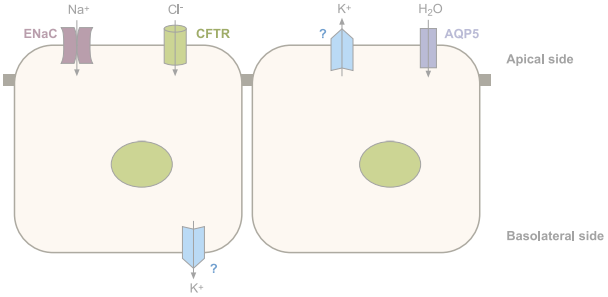
Figure 1b: Ion channels in lung alveolar epithelial cells.
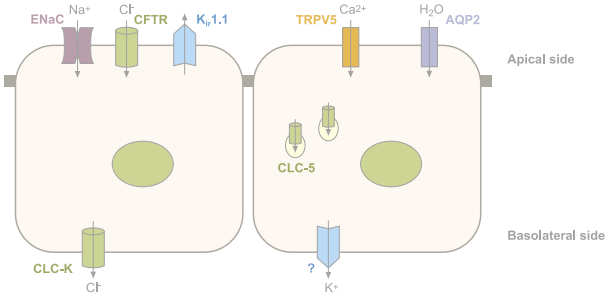
Figure 1c: Ion channels in renal epithelial cells. Schematic representation of the main ion channels in epithelial cells. Ubiquitous channels have been omitted. Vectorial Cl- transport in the colon through CFTR (absorption or secretion) varies according to the cell type.
Water Channels (Aquaporins) in Epithelial Tissue
Although water can cross the lipid bilayer by simple diffusion, this process is relatively slow. Tissues in which high permeability to water is necessary such as renal, lung and secretory epithelia express specialized water channels: the aquaporins. Eleven mammalian aquaporins (AQP0-AQP10) have been described so far with broad distribution in diverse epithelial cell membranes where they serve to regulate water reabsorption and body fl uid homeostasis. Some of the aquaporins have broad epithelial tissue distribution such as AQP1, AQP3 and AQP4 whereas others such as AQP2, AQP5 and AQP6 have a more restricted distribution.42 Only the AQP2 water channel will be considered here.
AQP2
AQP2 is found almost exclusively in renal collecting duct principal cells. This nephron segment is where the final volume and concentration of urine is determined. However, the collecting duct is permeable to water only in the presence of the antidiuretic hormone vasopressin. It was found that at basal conditions AQP2 expression in the collecting duct cells is mainly restricted to intracellular vesicles. Binding of vasopressin to its receptor, results in increased levels of cAMP that activate PKA, which in term directly phosphorylates AQP2 on serine 256. The phospho-AQP2-containing vesicles are then translocated to the apical membrane with a concomitant increase in water permeability. The water that enters the cells via apical AQP2 exits them via AQP3 and AQP4 that are constitutively expressed in the basolateral membrane.
When vasopressin is removed, AQP2 is reinternalized and the water permeability returns to baseline42, 43. The importance of the vasopressin-regulated water absorption mechanism is underscored by the identification of individuals with a genetic disease that can be ascribed to mutations in the AQP2 gene. Hereditary nephrogenic diabetes insipidus (NDI) is a rare disorder that results in the excretion of large volumes of undiluted urine. Two classes of mutations have been identified: mutations with recessive and mutations with dominant inheritance patterns. Recessive mutations cause misfolding and retention of the proteins in the ER and therefore no viable AQP2 expression. Dominant mutations on the other hand, produce proteins with trafficking defects and since the functional aquaporin channel is a tetramer, this kind of mutation will produce a dominant-negative effect in AQP2 translocation thus explaining the dominant pattern of inheritance44, 45.
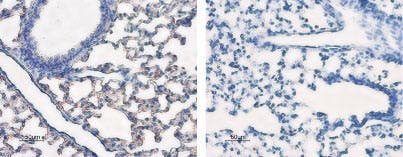
Expression of Aquaporin 5 in rat lung. Rat lungs were stained with Anti-Aquaporin 5 Antibody (#AQP-005) (left panel). The strong staining in alveoli (red-brown) is consistent with type I pneumocytes (arrows). Counterstain of cell nuclei appears blue. A negative control is shown in the right panel.
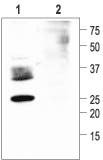
Western blotting of rat kidney membranes:
1. Anti-Aquaporin 2 Antibody (#AQP-002) (1: 200).
2. Anti-Aquaporin 2 Antibody, preincubated with a negative control antigen.
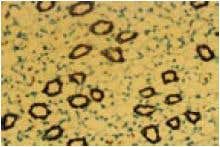
Expression of Aquaporin 2 in rat kidney. Immunohistochemical staining of Aquaporin 2 with Anti-Aquaporin 2 Antibody (#AQP-002) in rat kidney. The counterstain was methylene blue.
Conclusions
It is abundantly clear that ion channels play pivotal roles in epithelial function and hence whole body metabolism and homeostasis. Despite that, development of drugs that specifically target epithelial ion channels has been lagging. Most efforts have been directed towards increasing or restoring the Cl- conductance in CFTR-defective CF patients, whereas ENaC has been also proposed as a target for both CF and hypertension conditions. Clearly, in the next few years as the molecular identification of specific ion channels and our understanding of their biological function expand, we will see further efforts directed at the development of drugs targeting these channels.