The ENaC/degenerin channel superfamily comprises channels that are not voltage-gated but do involve the passage of ions, namely Na+. These channels are involved in many physiological processes, implicating them therefore in various pathophysiological diseases. Amiloride, mainly an ENaC channel inhibitor, Psalmotoxin-1 and APETx2, two peptide toxins isolated from two different venoms, inhibitors of ASIC1a and ASIC3 channels are commonly used to modulate their activity. This brief paper reviews some of the recent development regarding these channels by citing the use of Alomone Labs products.
Structure and Localization
ENaC channels are composed of three structurally related subunits: α, β, and γ which mostly coassemble as heteromeric structures in order to form functional channels. Structurally, channels belonging to the ENac/ Degenerin superfamily have short N- and C-termini and a large extracellular loop. In addition, strong evidence suggests that furin proteases cleave their extracellular loop for full channel activation. These channels are highly sensitive to amiloride and are hence commonly termed amiloride-sensitive channels.
ENaCs were initially detected in the kidney collecting duct where they are important for Na+ reabsorbtion, a task required for the concentration of urine25. Since, ENaCs have been identified in several other tissues including vascular smooth muscle, neurons, pancreas, testis, ovary, heart and lung23. Recently, immunohistochemical studies with Anti-ENaC β (SCNN1B) (extracellular) Antibody (#ASC-019) detected ENaCβ in the periodontal ligament which receives innervation from sensory neurons originating from the trigeminal ganglion (TG) and trigeminal mesencephalic nucleus13. ENaCs are also expressed in many tissues ranging from epithelial and endothelial cells, taste cells and various brain regions6.
ASIC channels belong to the ENaC/Degenerin superfamily and are protongated ion channels which are not voltage-dependent. Six different genes encode the different ASIC subunits and splice variants: ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3 and ASIC4. The topology of ASIC channels is predicted on the basis of that of ENaCs. However, different computational programs have depicted different structures for the ASIC channels. In a study, Anti-ASIC2a Antibody (#ASC-012) was used to show that ASIC2a is first and foremost a glycosylated protein as shown with endo H treatment. In addition, immunocytochemistry studies elegantly prove that the N-terminus of ASIC2a is intracellular as cell permeabilization is required in order to detect the channel at its N-terminus28 (Figure 1).
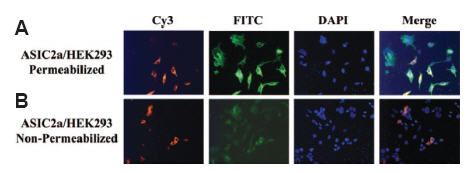
Anti-ASIC2a Antibody(#ASC-012) is targeted against amino acids 1-20. A) Permeabilized HEK293 cells expressing ASIC2a are immunopositive for ASIC2a (FITC-labeled secondary antibody) and tubulin (Cy3-labeled secondary antibody). B) When the same cells are not permeabilized, low levels of ASIC2a are present.
Adapted from reference 28 with permission of American Society for Biochemistry and Molecular Biology.
ASIC channel currents could be inhibited by natural peptides isolated from different venoms: Psalmotoxin-1 (PcTx-1) isolated from Psalmopoeus cambridgei tarantula venom, an inhibitor of ASIC1a and APETx2 isolated from Anthopleura elegantissima sea anemone, which inhibits ASIC3. Indeed, it was recently shown that Psalmotoxin-1 (#STP-200) has a very low effect on ASIC1b re-confirming that Psalmotoxin-1 is specific for ASIC1a30.
These channels are expressed in the brain and in the nervous system18 but have also been found in the retina, testes, and other tissues8,15 where they are involved in many physiological processes including nociception33, learning and memory35, mechanosensation27 among others.
Protein Modification and Protein Interaction
Channel trafficking and activity could be regulated by protein-protein interactions and also by post translation modifications of the channel. ASIC channels sense protons and interact with proteins (namely proteases) and other ions such as Zn2+ and Ca2+ through their extracellular domains. The extracellular loop of ASIC1a undergoes cleavage by extracellular trypsin (a serine protease) which is responsible for shifting the pH dependence of the channel activation and inactivation to more acidic pH26. A recent study demonstrated a correlation between the cleavage of ASIC1a and its functional outcome34. Mutagenesis studies show that trypsin cleaves ASIC1a in the N-terminus region of the extracellular loop between domains important for the inhibition of the channel by Psalmopeus cambridgei venom. Western blot analysis using Anti-ASIC1 Antibody (#ASC-014) showed that under control conditions, both ASIC1a and 1b, individually expressed in Xenopus oocytes, were detected as a single band (Figure 2) and that following cleavage with trypsin, ASIC1a but not ASIC1b migrated to a lower molecular weight position indicating that the channel was cleaved by the protease. Constructing chimeric proteins between ASIC1a and ASIC1b identified a region in ASIC1a sensitive to trypsin cleavage34.
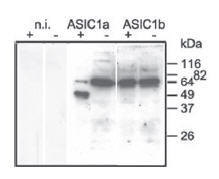
Xenopus oocytes were incubated with trypsincontaining extracellular solution (“+” if trypsin was present in the solution or “-” if trypsin was absent) for 5 min. n.i., non-injected oocytes. Cell surface proteins were biotinylated and ASIC1 was detected using Anti-ASIC1 Antibody(#ASC-014).
Adapted from reference 34 with permission of American Society for Biochemistry and Molecular Biology.
The intracellular N- and C-termini contain residues that can modulate channel activity via post-translational modifications or protein-protein interactions. Parkin is a 52 kD protein expressed in various tissues and acts as an E3 ubiquitin ligase29. Protein ubiquitination is a complex process which often leads to the degradation of the client protein via the 26S proteosome multimeric protein complex12. However, it is clearly understood that protein ubiquitination can also take place as a reversible post-translational modification, part of a cell signaling cascade. The discrimination of the pathway a ubiquitinated protein undertakes relies on the length of ubiquitin molecules added to the protein. Ubiquitinated proteins can function in a plethora of cellular activities, including signal transduction, transcription and membrane trafficking24. Parkin is involved in the monoubiquitination of PICK1, a PDZ protein known to interact with neurotransmitter receptors,transporters and ion channels20. PICK1, activated by PKC is also a positive regulator of ASIC channel activity3. Whether monoubiquitinated PICK1 has any effect on ASIC currents was addressed16. Results suggest that Parkin inhibits the ability of PICK1 to potentiate the activity of ASIC2a through ubiquitination. The depleted ASIC2a currents were not a result of decreased protein levels as Anti-ASIC2a antibody detected constant protein levels in the presence and absence of Parkin. As PICK1 regulates ASIC activity, the possibility of direct channel regulation by PKC was assessed (since PKC activates PICK1). Three consensus phosphorylation sites are present in ASIC1b. Mutations in these consensus sites were generated and protein levels of each mutant were monitored using Anti-ASIC1 in western blot analysis and the effect of each mutation was tested in eletrophysiological recordings. While two different mutated sites did affect ASIC1 current recordings, none of the mutants demonstrated effects on the expression of the channel5.
ASIC1a activation may be a major factor in events leading to neuronal damage. Using a yeast two hybrid screening system, p11, a member of the S100 small phospholipid and Ca2+ binding protein family was found to bind ASIC1a and involved in the trafficking of ASIC1a and other channels to the plasma membrane. This interaction is specific to ASIC1 as p11 did not interact with other ASIC channels. In order to show that these two proteins interact in vivo, mouse dorsal root ganglia (DRGs) were used to immunoprecipitate the two proteins. An anti-p11 antibody was used to immunoprecipitate the protein from DRGs and Anti-ASIC1 was used to show that ASIC1 is in fact co-immunoprecipitated with p11. ASIC1 channel was not detected in immunoprecipitates from ASIC1 knock out mice DRGs revealing that the interaction is specific and authentic10. p11 has also been attributed to pain sensation19. In order to fully investigate its role in nociception, p11 was knocked out in mouse DRGs. As ASIC1 is also involved in nociception, its protein levels were measured in p11 knocked out DRGs using Anti-ASIC1 in western blot analysis. Levels of the channel remained unchanged in these cells. However, the possibility remains that perhaps ASIC1 is expressed in other membrane compartments and not targeted to the plasma membrane11.
Increasing evidence indicate that ASICs and ENaCs are co-expressed in various tissues and cell types. ASIC1 and ASIC2 were used to investigate the possibility that ENaCs and ASICs may also form heteromeric structures23. ASIC1 and ASIC2 were co-transfected with their ASIC counterpart (i.e. ASIC1 with ASIC2) or cotransfected with a fluorescently tagged ENaC subunit. ASIC channels were immunoprecipitated with their respective Alomone Labs antibody and rather than running the immunoprecipitate on an SDS-PAGE gel, the protein A conjugated beads with the immune-complex were imaged by a confocal microscopy in order to determine the immunofluorescent complex23 (Figure 3). In every combination tested, yellow fluorescence (ENaC detection) was detected. Control experiments did not show yellow fluorescence, indicating that Anti-ASIC1 and Anti-ASIC2a antibodies do not cross-react with other ASICs or with ENaC channels. The study further shows that these heteromeric structures exhibit electrophysiological properties different from homomeric structures.
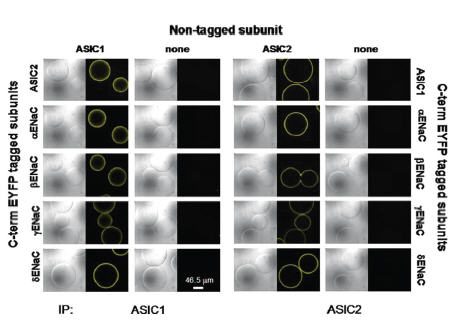
ASIC1 or ASIC2 were cotransfected, or not with their ASIC counterpart or with different ENaC subunits tagged to EYFP, yellow fluorescent probe. ASIC1 was immunoprecipitated with Anti-ASIC1 Antibody (#ASC-014), (left panels). Co-immunoprecipitation demonstrates intermolecular interactions between ASIC1 and ENaC as well as ASIC1 with ASIC2 using differential interference contrast and yellow fluorescence imaging of immunocomplexed agarose beads. The same was done for ASIC2a using Anti-ASIC2a Antibody(#ASC-012), (right panels).
Adapted from reference 23 with permission of American Society for Biochemistry and Molecular Biology.
Adenosine-monophosphate-activated kinase (AMPK) is a sensor for low cellular energy states by detecting the ATP/ADP ratio. It mainly has a protective role by inactivating ATP-consuming processes and activating ATP-generating pathways. The activity/conductance of ENaCs is inhibited by AMPK9. A recent study showed that AMPKα1 is the isoform crucial for ENaC regulation in various Na+ absorbing epithelia in vivo2. AMPK controls ENaC by facilitating ENaC ubiquitination by Nedd4-2 (an ubiquitin ligase) and its subsequent internalization. In fact AMPKα1-/- mice have increased Na+ absorption via ENaCs which is paralleled by an increase in ENaC expression in colon, kidney and airways as determined in immunohistochemistry studies using Anti-ENaC β (SCNN1B) (extracellular) Antibody.
Pathophysiology
In the central nervous system, gliomas are the most common type of tumors originating from astrocytes21. These tumors are divided into four major groups, based on how malignant they are. Glioblastoma multiforme (GBM), grade IV, is the most potent of the four groups. Grade IV type gliomas occur most frequently and have the worst prognostic. Glioblastomas are known to express amiloride-sensitive currents as opposed to astrocytes or less invasive gliomas. These currents are an attractive therapeutic approach as their inhibition decreases migration and growth of these cells. The currents measured in high grade glioma cells are composed of a mixture of both ASIC and ENaC subunits17. In fact, ASIC channels (namely ASIC1) are expressed to higher degrees compared to ENaCs in a glioma cell line when compared to normal astrocytes. In order to study the physiological significance of each channel type, knocked-down cells were generated. ASIC1 knockdown cells showed that the channel expression decreased by 60%17 as shown using Anti-ASIC1 antibody (Figure 4). Currents in these cells were also abolished similarly to those treated with Psalmotoxin-1. In this study, the authors further showed that ASIC1 and ENaCs could be immunoprecipitated, using Anti-ASIC1 antibody, strongly suggesting that the two proteins physically interact under physiological conditions.
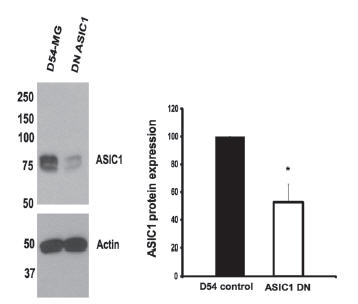
Western blot analysis of D54-MG gliblastoma lysates using Anti-ASIC1 Antibody(#ASC-014). D54-MG cells transfected with the knockdown construct shows a 50-60% decrease in the expression of ASIC1.
Adapted from reference 17 with permission of American Society for Biochemistry and Molecular Biology.
Through a long line of studies and results, it is hypothesized that ASIC2 does not reach the plasma membrane and its non-functionality is the cause for the constitutive inward Na+ currents observed in high grade gliomas. Indeed, it was found that both ASIC1 and ASIC2 are expressed in normal and high grade gliomas in western blot analysis using Anti-ASIC1 and Anti-ASIC2a antibodies. However, only ASIC1 seems to reach the plasma membrane while ASIC2 is retained in intracellular compartments, most likely the Golgi. Hsc70, a constitutively expressed heat shock protein, was thought to be involved in ASIC2 trafficking to the plasma membrane, since this protein is responsible for trapping ΔF508-CFTR mutant and preventing the misfolded channel from reaching the plasma membrane. The main cellular functions for Hsc70 are its binding to misfolded proteins and the ultimate recruitment of the ubiquitin-proteosome for the degradation of misfolded proteins7. As Hsc70 could also bind different ion channels including K+ channels and CFTR and mediate their trafficking, its possible involvement regarding ASIC2 trafficking was also studied32. Data show that Hsc70 is overexpressed in high glioma cells and immunoprecipitation studies using Anti-ASIC2a show that ASIC2 and Hsc70 interact with each other. Downregulating Hsc70 is also associated with increased localization of ASIC2 at the plasma membrane32. When ASIC2 is forced to reach the plasma membrane, the glioma cells cease to migrate and proliferate, strongly suggesting that ASIC2 plays an important role in these processes, probably by recruiting additional factors important for downregulating tumor progression31.
Tissue acidosis is an important characteristic of tumors. The response of adenoid cystic carcinoma cells (ACC) to acidic solution was studied using whole cell patch clamp recordings. In addition, western blot analysis using Anti-ASIC2a and Anti-ASIC3 Antibody (#ASC-018) antibodies demonstrated that these two channels are expressed in these tumor cells. Furthermore, these channels are also overexpressed in ACC tissue samples. In addition to high grade gliomas, ASICs may also play a role in other tumor cells, namely ACC where they might sense and respond to changes in extracellular pH in order to adapt to the metabolic activity38.
ASIC3 seems to be essential for the development of mechanical hyperalgesia following muscle insult as knockout mice do not develop mechanical hyperalgesia of the paw after the induction of inflammation (by either acid or carrageenan injections). In a recent study the role of ASIC3 in mechanical hyperalgesia associated with joint inflammation, as well as the expression of ASIC3 in primary afferent fibers innervating the knee joint were investigated. Behavioral studies using ASIC3+/+ and ASIC3-/- mice indicate that ASIC3 is mostly required for secondary hyperalgesia but not primary hyperalgesia. Immunohistochemistry studies using Anti-ASIC3 antibody demonstrate that ASIC3 is in fact localized to primary afferent fibers innervating the knee joint, important for increasing the input to the spinal cord a process required for secondary hyperalgesia14.
Symptomatic gastro-oesophageal reflux disease (GORD) and related syndromes are mainly characterized by increased acid reflux in the esophagus. At the molecular level, acid sensors must be present and take part in the detection of the acidic milieu. ASIC1, 2 and 3 are all detected in the esophageal epithelium, and are observed in the prickle cell in the esophageal mucosa using their respective Alomone Labs antibodies1 (Figure 5). In addition, both TRPV1 and ASIC3 are also detected in the muscularis mucosa using Anti-TRPV1 (VR1) Antibody (#ACC-030) or Anti-Rat TRPV1 (VR1) (extracellular) Antibody (#ACC-029) and Anti-ASIC3 Antibody. Along with the detection of these acid sensors, the alteration of these targets may be the cause why GORD related patients suffer from extreme acid reflux1.
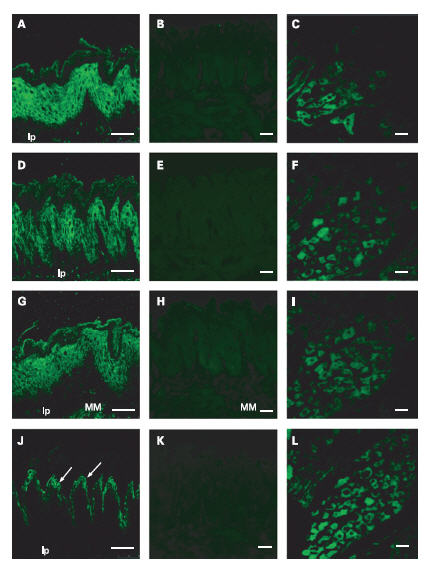
ASIC1 detected with Anti-ASIC1 Antibody(#ASC-014), (A), ASIC2a detected with Anti-ASIC2a Antibody (#ASC-012), and ASIC3 detected with Anti-ASIC3 Antibody(#ASC-018) are expressed in prickle cells. ASIC3 is also expressed in the muscularis mucosa (MM), (G). TRPV1 immunoreactivity using Anti-TRPV1 (VR1) Antibody(#ACC-030) or Anti-Rat TRPV1 (VR1) (extracellular) Antibody (#ACC-029) is observed in the granular cells (arrows) in the stratum granulosum ( J). Preabsorbed antibody with the immunizing peptide was done for ASIC1 (B), ASIC2a (E), ASIC3 (H), and TRPV1 (K). Positive control staining in dorsal root ganglia was done for ASIC1 (C), ASIC2 (F), ASIC3 (I) and TRPV1 (L). lp, lamina propria mucosa. Scale bar, 50 μm.
Adapted from reference 1 with permission of BMJ Group.
20-25% of today’s population is affected by hypertension37. Hypertension is also characteristic of pseudohypoaldosteronism type II (PHAII), an autosomal dominant disorder. Individuals suffering from hypertension in PHAII are molecularly characterized as having increased Na+ absorption and/or dysfunctional K+ channels in the kidneys. Recently, mutations in the with no lysine kinase 1 (WNK1) and 4 (WNK4) genes encoding serine/threonine kinases, were shown to be the cause for PHAII pathogenesis36. Following preliminary characterization of mutated Wnk knockin mice which develop hypertension like PHAII patients, the involvement of ENaCs and ROMK1 (Kir1.1, KCNJ1), two important channels for Na+ reabsorption and K+ in the cortical collecting ducts (CCD) in PHAII was investigated. Western blot and immunohistochemical studies using Anti-ENaC β (SCNN1B) (extracellular) Antibody, Anti-KCNJ1 (Kir1.1) Antibody (#APC-001) and Anti-KCNMA1 (KCa1.1) (1097-1196) Antibody or Anti-KCNMA1 (KCa1.1) (1184-1200) Antibody (#APC-021 or #APC-107) show that EnaCβ and KCa1.1 (Maxi K) are elevated while ROMK1 remains unchanged in knocked in mice. These effects however, seem to be secondary and the main cause appears to be the increased activity of the Na-Cl cotransporter (NCC) through the activation of OSR1/SPAK-NCC kinase cascade37.
It is well established that in sensory neurons, ASIC channels are associated with pain sensation22. ASIC1a and the two ASIC2 isoforms are expressed in the spinal cord; however data on the expression of ASICs in the spinal cord neurons, the first central neurons to integrate and modulate noxious signals generated by the peripheral nociceptors, are limited. After validating that ASIC-dependent currents are indeed expressed in dorsal spinal neurons, their expression was monitored using both in situ hybridization and immunohistochemistry4. Fluorescent labeling of ASIC1a in in situ hybridization shows clear colocalization with ASIC2a detected by immunohistochemistry with Anti-ASIC2a Antibody, in different spinal lamina sections, consistent with electrophysiological data. In addition, the electrophysiological data strongly point to a heteromerization between these two channel isoforms.
References
- Akiba, Y. et al. (2008) Gut 57, 1654.
- Almaca, J. et al. (2009) Pflugers Arch. 458, 713.
- Baron, A. et al. (2002) J. Biol. Chem. 277, 50463.
- Baron, A. et al. (2008) J. Neurosci. 28, 1498.
- Bashari, E. et al. (2009) Am. J. Physiol. 296, C372.
- Benos, D.J. and Stanton, B.A. (1999) J. Physiol. 520 Pt 3, 631.
- Bercovich, B. et al. (1997) J. Biol. Chem. 272, 9002.
- Brockway, L.M. et al. (2002) Am. J. Physiol. 283, C126.
- Carattino, M.D. et al. (2005) J. Biol. Chem. 280, 17608.
- Donier, E. et al. (2005) J. Biol. Chem. 280, 38666.
- Foulkes, T. et al. (2006) J. Neurosci. 26, 10499.
- Hershko, A. and Ciechanover, A. (1998) Annu. Rev. Biochem. 67, 425.
- Hitomi, Y. et al. (2009) Biomed. Res.-Tokyo. 30, 113.
- Ikeuchi, M. et al. (2008) Pain 137, 662.
- Ishibashi, K. and Marumo, F. (1998) Biochem. Biophys. Res. Commun. 245, 589.
- Joch, M. et al. (2007) Mol. Biol. Cell. 18, 3105.
- Kapoor, N. et al. (2009) J. Biol. Chem. 284, 24526.
- Krishtal, O. (2003) Trends Neurosci. 26, 477.
- Ling, Q. et al. (2004) J. Clin. Invest. 113, 38.
- Madsen, K.L. et al. (2005) J. Biol. Chem. 280, 20539.
- Maher, E.A. et al. (2001) Genes Dev. 15, 1311.
- McCleskey, E.W. and Gold, M.S. (1999) Annu. Rev. Physiol. 61, 835.
- Meltzer, R.H. et al. (2007) J. Biol. Chem. 282, 25548.
- Mukhopadhyay, D. and Riezman, H. (2007) Science 315, 201.
- Palmer, L.G. and Frindt, G. (1986) Proc. Natl. Acad. Sci. U.S.A. 83, 2767.
- Poirot, O. et al. (2004) J. Biol. Chem. 279, 38448.
- Price, M.P. et al. (2000) Nature 407, 1007.
- Saugstad, J.A. et al. (2004) J. Biol. Chem. 279, 55514.
- Shimura, H. et al. (2000) Nat. Genet. 25, 302.
- Springauf, A. and Grunder, S. (2010) J. Physiol. 588, 809.
- Vila-Carriles, W.H. et al. (2006) J. Biol. Chem. 281, 19220.
- Vila-Carriles, W.H. et al. (2007) J. Biol. Chem. 282, 34381.
- Voilley, N. et al. (2001) J. Neurosci. 21, 8026.
- Vukicevic, M. et al. (2006) J. Biol. Chem. 281, 714.
- Wemmie, J.A. et al. (2002) Neuron 34, 463.
- Wilson, F.H. et al. (2001) Science 293, 1107.
- Yang, S.S. et al. (2007) Cell Metab. 5, 331.
- Ye, J.H. et al. (2007) Biochem. Biophys. Res. Commun. 355, 986.