K+ channels are the largest and most diverse superfamily of ion channels. They are ubiquitously expressed in both excitatory and non-excitatory cells where they play a role in an assortment of different physiological functions such as cell proliferation and apoptosis, neuro- and cardio-protection, neuronal excitability, muscle contraction and salt secretion. Among this diversity the Ca2+-dependent K+ channel (KCa) subfamily is unique in that they integrate changes in intracellular Ca2+ concentrations with membrane excitability in both neuronal and non-neuronal cell types.
Introduction
The KCa channel subfamily of K+ channels can be classified into three groups based on their biophysical characteristics and single channel conductance (see Table for the current nomenclature). The large conductance (100-300 pS) KCa group includes the KCa1.1 (KCNMA1) channel, the intermediate conductance (25-100 pS) consists of the KCa3.1 channel and the small conductance (2-25 pS) group comprises the KCa2.1, KCa2.2 and KCa2.3 channels. The large conductance KCa1 group probably also includes the products of three other novel genes: KCa4.1 (or KCNT1, Slack), KCa4.1 (KCNT2) and KCa5.1 (KCNMC1, slo-3), some of which likely function as Na+-dependent K+ channels and will not be further discussed in this review.
All KCa channels open in response to an increase in intracellular Ca2+ in the micromolar range while the KCa1.1 channel is the only one that can also be activated by membrane depolarization. The KCa1.1 is also unique in its topology, which includes seven transmembrane domains with an extracellular N-terminus and differs from that of the other KCa channels that resemble the structure of the voltage-gated K+ channels (six transmembrane domains with intracellular N- and C-termini). The functional channel of all the KCa family members is a multimeric protein composed of four pore-forming subunits with (in the case of the KCa1.1 channel) an occasional auxiliary (β) subunit.
KCa channels are expressed in virtually all cells where they integrate cellular metabolism with membrane excitability thus contributing to various and diverse physiological outcomes. In this review we will summarize the biophysical characteristics of these channels and some of their physiological functions.
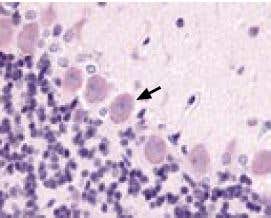
Figure 1. Expression of KCNMA1 (KCa1.1) in rat cerebellum. Immunohistochemical staining of KCNMA1 channel with Anti-KCNMA1 (KCa1.1) (1097-1196) Antibody (#APC-021) in rat cerebellum. Picture showing the Purkinje layer. Note that Purkinje cells (arrow) show an intense staining. Reaction product is pale red and counterstain is hematoxylin.
Immunohistochemistry data provided by LifeSpan Biosciences, Seattle, USA.
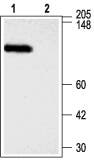
Figure 2. Western blotting of rat brain membranes:
1. Anti-KCNMA1 (KCa1.1) (1097-1196) Antibody (#APC-021) (1:300).
2. Anti-KCNMA1 (KCa1.1) (1097-1196) Antibody, preincubated with the negative control antigen.
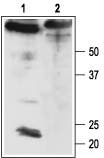
Figure 3. Western blotting of rat brain membranes with Anti-sloβ4 Antibody:
1. Anti-sloβ4 Antibody (#APC-061) (1:200).
2. Anti-sloβ4 Antibody, preincubated with the negative control antigen.
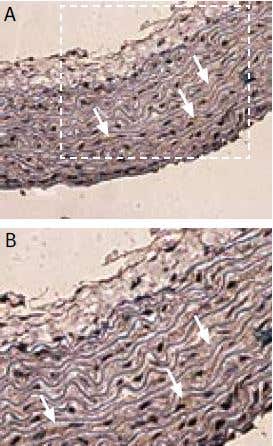
Figure 4. Expression of sloβ1 in rat pulmonary artery. Immunohistochemical staining of sloβ1 with Anti-sloβ1 (KCNMB1) Antibody (#APC-036) in rat pulmonary artery smooth muscle cells. A) Transversal section of the pulmonary artery. Arrows show strongly stained myocytes. B) Enlargement. DAB product is brown and the counterstain is Cresyl Violet.
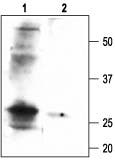
Figure 5. Western blotting of rat smooth muscle lysate:
1. Anti-sloβ1 Antibody (#APC-036) (1:200).
2. Anti-sloβ1 Antibody, preincubated with the negative control antigen.
1. KCa1.1: Modulating Smooth Muscle Tone and Neurotransmitter Release
General Considerations
The KCa1.1 channel is by far the most studied of the KCa subfamily. The channel is ubiquitously expressed in almost all cell types where it participates in an astounding array of physiological functions. It was first characterized in Drosophila as the slowpoke channel and later identified in mouse and humans1. Given the fact that native currents of the KCa1.1 channel with very different properties were known at the time, it was surprising that only one gene corresponding to the channel had been identified. Moreover, the KCa1.1 channel gene is remarkably conserved among different species in mammals, suggesting that there is evolutionary pressure to maintain its optimized function1,2. The different native channel properties found in various tissues can two transmembrane domains, an extracellular loop and cytoplasmic N- and C-termini (see Table) and have different expression patterns. The first regulatory subunit (now termed sloβ1) was identified in smooth muscle preparations on the basis of its high affinity for Charybdotoxin (ChTX) a known peptide blocker of KCa1.1 (see Table)3,4. As a whole, the four regulatory subunits increase the sensitivity of the pore-forming KCa1.1 subunit to Ca2+ and voltage and they can also change its pharmacology and/or act as receptors for drugs.
The physiological role of the β subunits will be discussed in more detail below. As mentioned above, alternative splicing of the KCa1.1 that accounts for physiological differences has been identified in various tissues such as adrenal chromaffin cells, brain and human gliomas5-7.
At least six sites for alternative splicing have been identified in the mouse KCa1.1 transcript. Functional properties of the different splice variants are only starting to emerge but changes in Ca2+ sensitivity and/or slowed channel activation has been described for some of the different variants. In addition, the KCa1.1 can be modulated by a large array of cellular signaling pathways including protein phosphorylation and/or dephosphorylation, G-proteins and nitric oxide8-10. Regulation of the KCa1.1 channel by protein phosphorylation has been more thoroughly addressed. Although some tissue specific differences have been detected, for the most part channel phosphorylation via protein kinase A (PKA) and protein kinase G (PKG) seems to stimulate channel activity in smooth muscle by altering the responsiveness of the channel to Ca2+8. In addition, several proteins have been identified that can associate with the KCa1.1 channel and include Syntaxin 1A, β2 adrenergic receptors and β-catenin11-13. These proteins can affect the distribution of the channel within the cell; thus changing the current modulation and subsequent physiological outcome. It seems conceivable that as better biochemical and molecular tools became available the list of proteins found to be associated with the KCa1.1 channel will steadily grow.
Physiological Significance
As the complex modulation of the KCa1.1 channel would suggest, the activity of the channel profoundly influences several physiological functions in various tissues. That is not surprising if one considers that the channel sits at the crossroads of two pathways that profoundly affect all cell function: changes in cytosolic Ca2+levels and membrane potential. We will briefly consider two fields in which the importance of the KCa1.1 channel at the physiological level has been more extensively studied: modulations of smooth muscular tone and Ca2+-dependent neurotransmitter release. KCa1.1 channels have been identified in a variety of smooth muscle preparations including vascular, urinary bladder, uterine and others. In all smooth muscle preparations, the channel appears to be functionally coupled with the sloβ1 auxiliary subunit that is exclusively expressed in smooth muscle. Simply put, smooth muscle cells contract as a result of an increase in the intracellular Ca2+ concentration. This Ca2+ increase activates the Ca2+-calmodulin-dependent protein kinase, which initiates a biochemical cascade that eventually results in muscular contraction. The intracellular Ca2+ increase is mediated by the opening of ryanodine-receptors in the sarcoplasmic reticulum (Ca2+ sparks) and/or the opening of voltage-dependent Ca2+ channels (VDCCs) in the plasma membrane. Both the rise in intracellular Ca2+ and the membrane depolarization (that opens the VDCCs) will activate the KCa1.1 channel that responds with an efflux of K+ and a concomitant hyperpolarization of the cell membrane potential. This in turn, will close the membrane VDCCs thus effectively working as a negative feedback mechanism on contraction and inducing muscle relaxation. Indeed, inhibition of KCa1.1 channel activity in vascular smooth muscle preparations with Iberiotoxin (a specific channel blocker, see Table) has been shown to induce membrane depolarization and vasoconstriction14. Conversely, KCa1.1 channel openers would act as relaxation factors of vascular smooth muscle, inducing membrane hyperpolarization and closure of Ca2+ channels. Indeed, several known endogenous vasodilators such as nitric oxide, atrial natriuretic factor, β adrenergic agonists, etc. produce their vasorelaxing effect by directly or indirectly (via activation of PKA and/or PKG) activating the KCa1.1 channel15, 16.
One of the most intriguing aspects of the involvement of the KCa1.1 channel in smooth muscle tone regulation is the involvement of the β1 auxiliary subunit. Studies with sloβ1 knockout mice have demonstrated that loss of this regulatory subunit produce hypertension and cardiac hypertrophy, probably because loss of this subunit decreases the Ca2+ sensitivity of the KCa1.1 channel, which can no longer respond to endogenous Ca2+sparks17,18. Conversely, a recent study showed that a commonly found gain-of-function sloβ1 variant has a protective effect against human diastolic hypertension. The sloβ1 variant does so by further increasing the apparent Ca2+ and voltage-activation sensitivity of the pore forming KCa1.1 channel19.
Although the widespread expression of the KCa1.1 channel in the nervous system has long been acknowledged, the functional role of these channels in the brain is largely unknown20. Similarly to their function in smooth muscle tone regulation, KCa1.1 channels are believed to function as a feedback inhibition mechanisms that initiates membrane repolarization and prevents additional Ca2+ entry through VDCC in neurons and hence further neurotransmitter release. Thus, the KCa1.1 channel is believed to act in the brain as an “emergency break” in situations that involve excessive depolarization and Ca2+ entry in pathological situations such as ischemia (reduced blood flow) or epilepsy21.
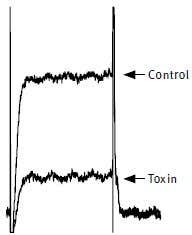
Figure 6. Inhibition of KCa1.1 channels by Charybdotoxin. The effect of 100 nM Charybdotoxin (#STC-325) on KCa1.1 (mSlo) expressed in Xenopus oocytes. Superimposed traces before and during perfusion of the toxin. Membrane holding potential was –100 mV, stepped every 15 s. to +20 mV for 100 ms. The vertical bar represents 0.1 μA for KCa1.1.
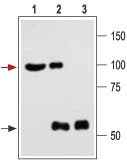
Figure 7. Immunoprecipitation of rat brain with Anti-KCNMA1 (KCa1.1) (1184-1200) Antibody:
1. Brain lysate.
2. Brain lysate immunoprecipitated with Anti-KCNMA1 (KCa1.1) (1184-1200) Antibody (#APC-107) (4 μg).
3. Brain lysate immunoprecipitated with pre-immune rabbit serum.
The upper arrow indicates the KCa1.1 channel while the lower arrow indicates the IgG heavy chain.
Western blotting was performed with Anti-KCNMA1 (KCa1.1) (1184-1200) Antibody.
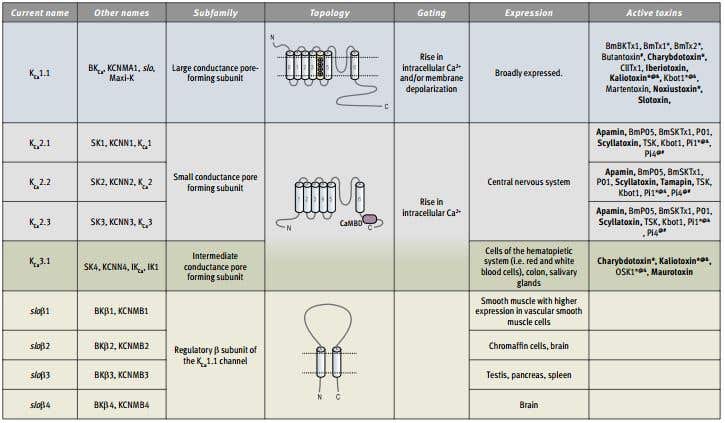
Table 1. The KCa Family Members and Auxiliary Subunits. Symbols denote other channels that are also blocked by the toxin.
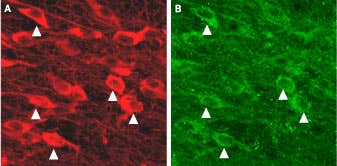
Figure 8. Expression of KCa2.3 in dopaminergic neurons (Mouse Substantia nigra). Immunohistochemical staining of KCa2.3 with Anti-KCNN3 (KCa2.3, SK3) (N-term) Antibody (#APC-025) (A) in mouse substantia nigra pars compacta. In (B), tyrosine hydroxylase staining shows dopaminergic neurons. Triangles point at cells with co-localization.
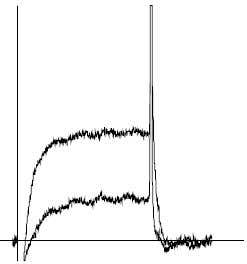
Figure 9. Inhibition of KCa1.1 channels by Iberiotoxin. KCa1.1 channel currents (5 mM K+) from Xenopus oocytes, before and during application of 100 nM Iberiotoxin (#STI-400). Holding potential, -100 mV, test potential, +50 mV for 100 ms. The vertical bar represents 0.1 μA.
2. KCa2: Modulating Neuronal Excitability
General Considerations
As mentioned above, the small conductance Ca2+-activated K+ (KCa2) channel subfamily comprises three highly homologous members: KCa2.1, KCa2.2 and KCa2.3 (see Table for the alternative terminology).22 They have a similar topology to the voltage-activated K+ (KV) channels but display only two positively charged amino acids at the S4 segment while KV channels typically display seven. This difference probably accounts for the observed voltage insensitivity of the KCa2 channels. KCa2 channels are therefore exclusively activated by increasing cytosolic Ca2+ concentration23. In fact, very low levels of intracellular Ca2+ (300-700 nM) can open the KCa2 channels very rapidly (5-15 ms). Hence, the KCa2 channels are highly sensitive and fast Ca2+sensors resembling other known Ca2+-binding proteins. This type of Ca2+-dependent activation is achieved by the constitutive binding of the KCa2 channels to calmodulin, a highly expressed Ca2+-binding protein via a calmodulin-binding domain situated at the cytoplasmic C-termini (see Table)24.
Several studies have detected heteromeric assembly of the KCa2 subunits (and even with the KCa3.1 subunit) in heterologous expression systems, although whether this occurs in native tissues is less clear23. Pharmacologically, the KCa2 channels are the only known targets of the bee venom toxin Apamin, with KCa2.1 being the least sensitive, KCa2.2 the most sensitive and KCa2.3 showing intermediate sensitivity25.
Physiological Significance
Based on their biophysical properties and central nervous system distribution, the KCa2 channels have long been implicated in the regulation of neuronal excitability. As mentioned above for the KCa1.1 channel, Ca2+enters neurons through VDCCs during an action potential (AP). Following the AP a membrane hyperpolarization occurs termed afterhyperpolarization (AHP), which dampens neuronal excitability. Several types of AHP are temporally distinguished, the fast (fAHP), the medium (mAHP) and the slow (sAHP) while the importance of each AHP depends on the type of neuron. KCa2 channels are believed to underlie the mAHP meaning that they are involved in the control of firing rate (the number of APs generated over a unit of time) and of firing pattern (the way the APs are distributed over time)26. These properties imply that in different neuronal populations, KCa2 activity will produce different outputs. For example, midbrain dopaminergic neurons have a prominent mAHP that is mediated by KCa2 channels (particularly KCa2.3) that control firing rate and subsequent dopamine secretion27. Since malfunction of these neurons is involved in several pathological disorders such as Parkinson’s disease and schizophrenia, modulators of the KCa2.3 channels have been proposed to be of therapeutic value in these diseases26.
KCa3.1: Modulating Lymphocyte Activation
General Considerations
The KCa3.1 channel is the lone member of the Ca2+-dependent K+ family of intermediate conductance. Ironically, this channel is the least studied of the KCa family even though it was the first one to be identified as the Ca2+-dependent K+ channel present in human erythrocytes where it is also known as the Gardos channel28. The channel has the same basic topology of the KCa2 subfamily and like them is constitutively bound to calmodulin. The channel is extremely sensitive to intracellular Ca2+ concentrations with an activation threshold of 200-300 nM29. The tissue distribution of the channel also varies substantially from that of the KCa2 subfamily. While the latter is expressed mainly in the central nervous system, the KCa3.1 channel is found exclusively in the periphery, in cells of hematopoietic origin, colon and salivary glands. Pharmacologically there are also marked differences, as the KCa3.1 channel is insensitive to apamin, the paramount channel blocker for the small conductance family. Instead, the channel can be blocked by the peptide toxins Charybdotoxin (that also blocks KCa1.1) and Maurotoxin (that also blocks KV1.2) (see Table).
Physiological Significance
Since its functional identification in human erythrocytes the physiological significance of the KCa3.1 channel has primarily been studied in cells of hematopoietic origin. In normal resting T-lymphocytes levels of the KCa3.1 channel are relatively low and it is believed that the voltage-dependent K+ channel KV1.3 is the main channel responsible for maintaining the cell membrane potential. However in activated T-lymphocytes the levels of KCa3.1 are markedly upregulated30. In order to be properly activated, that is capable of proliferation and differentiation into an effector T-lymphocyte, the cells must be able to maintain a sustained Ca2+ entry for at least a few hours. Because a high and steady Ca2+ entry would eventually dissipate the electrochemical force for additional Ca2+ entry, the opening of the KCa3.1 channels and the concomitant efflux of K+ would provide a hyperpolarization effect that would aid in the maintenance of the Ca2+ entry31. All this implies that specific blockers of the KCa3.1 channel would be able to prevent T-lymphocyte proliferation32. Indeed, several in vitro studies show that this is the case33,34. Moreover, recent studies have shown that in vivo blockage of the KCa3.1 channel can be useful in pathological situations that involve excessive T-lymphocyte-mediated activation. These include T-lymphocyte-mediated autoimmune diseases such as multiple sclerosis and T-lymphocyte-mediated inflammation among others35,36.
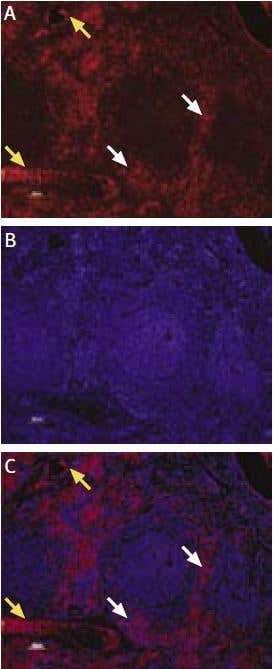
Figure 10. Expression of KCa3.1 in rat spleen. Immunohistochemical staining of KCa3.1 with Anti-KCNN4 (KCa3.1, SK4) Antibody (#APC-064) in rat spleen. (A) and (C) Secondary (activated) follicle in the spleen white pulp showing intense staining of Marginal Zone and Periarteriolar T-lymphocytes (white and yellow arrows, respectively); note that cells in the red pulp and B lymphocytes in the germinal center are not stained. (B) The counterstain is Hoechst 33324.
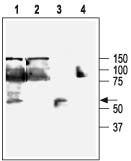
Figure 11 Western blotting of HEK-293-KCa3.1 (1, 2) and K562 (3, 4) cells:
1,3. Anti-KCNN4 (KCa3.1, SK4) Antibody (#APC-064) (1:200).
2,4. Anti-KCNN4 (KCa3.1, SK4) Antibody, preincubated with the negative control antigen.
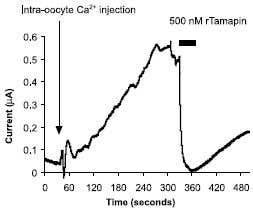
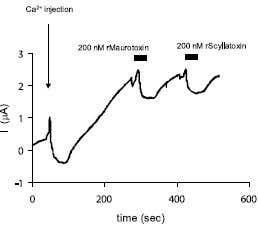
Figure 12. Inhibition of KCa2 K+ channels by Tamapin. Tamapin is a 31 amino acid peptidyl toxin, isolated from the venom of the Indian red scorpion, Mesobuthus tamulus. Activation of Tamapin (#STT-400) sensitive outward current by intracellular Ca2+ injection into Xenopus oocytes expressing SK2 (KCa2.2) channels. The arrow and vertical bar represents time of intracellular injection and period of toxin perfusion, respectively.
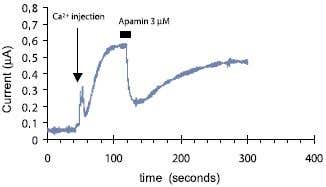
Figure 13. Inhibition of KCa2.2 (SK2) channels by Apamin. Voltage clamped whole oocyte current was recorded continuously under low Cl- concentration at 5 mV holding potential. At the indicated time 10 nl of 100 mM Ca2+ was injected into the oocyte and an outward current developed. A minute later 3 μM Apamin (#STA-200) was perfused into the bath, resulting in about 60% inhibition in the amplitude of this Ca2+ dependent current, which was nearly completely recovered upon toxin wash.