Neurodegenerative diseases take an enormous toll on society as well as on individuals. Very limited treatments for these diseases exist, since their molecular mechanisms are not well understood. Several ion channels, a group of membrane spanning proteins that regulate ion content and membrane potential in cells, have been implicated as important players in these diseases. While many Ca2+ conducting channels (CaV, P2X or glutamate receptors) may contribute to glutamate overload and excitotoxicity directly, K+channels often regulate the membrane potential controlling the Ca2+ signal indirectly. Below, we review the use of Alomone Labs’ products in research related to the role ion channels play in these diseases.
Sodium, potassium, and calcium channels, as well as the glutamate receptors NMDA and AMPA have all been implicated in various neurodegenerative diseases. One of the main factors in neurotoxicity is glutamate overload.1 Glutamate is the primary excitory neurotransmittor of the nervous system. Following glutamate release, postsynaptic responses occur through both metabotropic and ionotropic receptors. However, in 1957, excess glutamate was shown to be toxic. The term “excitoxicity” was coined to represent the neurotoxicity caused by excess of excitory neurotransmitters.2 It is now well accepted that a strong relationship exists between excessive Ca2+ influx and glutamate-triggered neuronal injury.3 The NMDA receptor in particular has been implicated in this process4 as well as the P2X7 receptor. P2X7 receptors are localized to the excitatory terminal s in the hippocampus, as shown by immunohistochemistry using Anti-P2X7 Receptor Antibody (#APR-004).5 Calcium overload can trigger many downstream neurotoxic cascades including activation of proteases, protein kinases, nitric oxide synthesase, calcineurins, which leads to increased production of toxin-reactive oxygen species, alteration in the cytoskelton, mitochondrial dysfunction, and activation of genetic signals leading to cell death.1 The pervasive involvement of Ca2+ in neuronal function suggested that altered Ca2+ homeostasis may be a fundamental mediator of age related changes in the nervous system.6
Alzheimer’s disease is a progressive, neurodegenerative disease characterized by loss of function and death of nerve cells in several areas of the brain, leading to loss of mental functions such as memory and learning. It is the most common cause of dementia. The neurodegeneration in Alzheimer’s disease is postulated to involve the loss of acetylcholine receptors from the basal forebrain cholinergic neurons (BCFN)7 , whose numbers are shown to decrease early in the disease process. In addition, potassium channels, specifically KV3.1 and KV2.1 have been implicated. Immunohistochemical techniques, using Anti-KV3.1b (KCNC1) Antibody (#APC-014) and Anti-KV2.1 Antibody (#APC-012) were used to investigate the expression of these channels and both were found to be expressed in the BCFN.7 Basal forebrain neurons are susceptible to changes associated with aging and related disfunctions, such as Alzheimer’s. The involvement of the smooth endoplasmic reticulum (SER) was investigated using the SER Ca2+ uptake blocker, Thapsigargin (#T-650), however it was not found to mediate the age-related changes.6
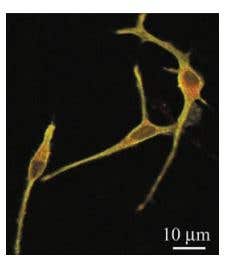
Figure 1. Membrane Localization of KV1.3 in Rat Microglial Cultures. Representative confocal immunofluorescence images of rat microglia showing colocalization of the membrane-delimited marker, OX-42, with KV1.3. OX-42 primary antibody was used with a biotinylated secondary antibody and FITC-conjugated streptavidin (green labeling), and Anti-KV1.3 (KCNA3) Antibody (#APC-002) primary antibodies were used with a Cy3-conjugated secondary antibody (red labeling). For each cell, the same confocal plane was used for acquisition, thus colocalization of channel and microglial membrane is represented by yellow regions in the merged images.
Adapted from Ref. # 11, with the kind permission of Dr. L.C. Schlichter of the Toronto Western Hospital and the Am. J.Physiol. Cell. Physiol.
The accumulation of plaques of amyloid beta (apeptide of 39–43 amino acids) is a hallmark of the disease and much effort has been expended to understand how these plaques interfere in the normal functioning of the brain. It has been suggested that these amyloid plaques form calcium-conducting ion channels that cause rapid neurodegeneration due to calcium overload.8 In addition, the plaques have been postulated to interfere with neuronal signaling via the nicotinic acetylcholine receptor in the early stages of the disease.7 The stimulatory effect of amyloid beta was partially dependent on voltage gated Ca2+ channels at low concentrations, as shown by the ability of a mixture of Ca2+ channel blockers, ω-Agatoxin-TK (#STA-530), ω-Conotoxin MVIIC (#C-150) and ω-Conotoxin GVIA (#C-300) to partially attenuate increases in Ca2+concentration in isolated hippocampal nerve endings in the rat.7 A-type K+ channels have been implicated in the onset of LTP in mammalian neurons, which is thought to underlie learning and memory, which are progresssively impaired in Alzheimer’s disease. To determine which channels underlie these currents in a drosophila model, specific blocker of the KV4.2 channel, Phrixotoxin-2 (#STP-710) and specific blocker of the Shaker channel α-Dendrotoxin (#D-350) were used. Treatment of cells with amyloid peptide altererd the kinetics of the current and caused a decrease in neuronal viability.9 Glutamate toxicity has also been postulated, as the NMDA receptor antagonist, memantine, has been approved for the treatment of Alzheimer’s.10
Damage by reactive oxygen species caused by the activation of microglia has been documented in Alzheimer’s disease. Several K+ channels have been shown to be involved in this respiratory burst by blocking current in cultured rat glia using the specific K+ channel blocker Agitoxin-2 (#STA-420).11 Blockage of the respiratory burst by Agitoxin-2 further demonstrated the involvement of K+ channels. Western blot analysis of microglial lysates using Anti-KV1.3 (KCNA3) Antibody (#APC-002), Anti-KV1.5 (KCNA5) Antibody (#APC-004) and Anti-KCNN3 (KCa2.3, SK3) (C-term) Antibody (#APC-103) and confocal imaging corroborated these findings. KV1.3 blockers Charybdotoxin (#STC-325), Agitoxin-2 and α-Dendrotoxin reduce neuron killing by microglia as shown by TUNEL analysis of neurons treated by activated microglia. KCa3.1 is highly expressed in microglia, as shown by immunobloting with Anti-KCNN4 (KCa3.1, SK4) Antibody (#APC-064) and has been shown to be a possible therapeutic target in acute and chronic neurodegenerative disorders.13
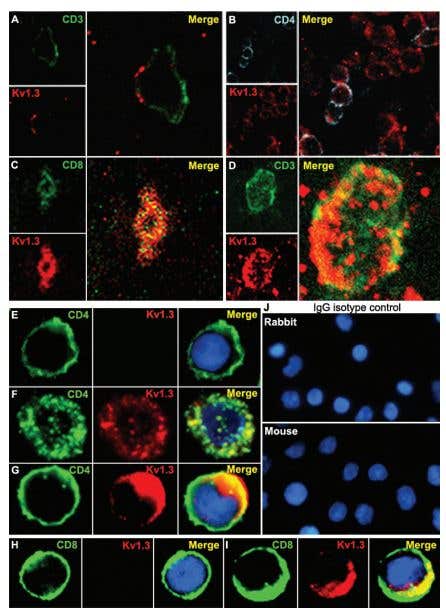
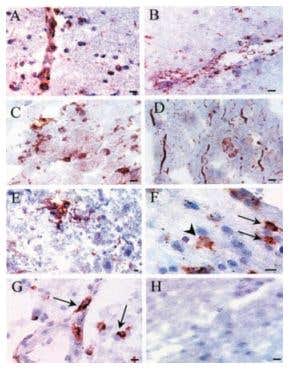
Figure 2. Immunohistochemical Staining of KV1.3 in Human MS Brain. Immunohistochemical staining of KV1.3 in human MS brain floating sections using Anti-KV1.3 (KCNA3) Antibody (#APC-002). Confocal microscopy of KV1.3 and CD3/CD4 in MS brain tissue. Confocal microscopic images of 20-µm floating sections of postmortem MS brain tissue. (A and B) Parenchymal infiltrate with positive CD3 and punctate KV1.3 staining in the membrane. (C)A cluster of lymphocytes stained positively for KV1.3 and CD4, as well as occasional CD8 (Inset). (D) CD3- cells showed extensive KV1.3 and CD3 colocalization in the membrane. Immunofluorescent staining of PB-derived T cells concentrated on a slide by cytospin showed membrane patterns of staining of CD4, CD8, and KV1.3. (E) Day 7-activated naive and TCM are CD4-, CCR7- (data not shown) and KV1.3-. (F) Chronically stimulated resting TEM were CD4-, CCR7- (data not shown) and expressed KV1.3 in the membrane but with minimal colocalization with CD4. (G) TEM that had been recently activated expressed increased amounts of KV1.3 and demonstrated more colocalization with CD4 similar to the brain tissue lymphocyte in D. (H) Naive CD8- T cells expressed no KV1.3 but, when chronically stimulated and activated (I), exhibited intense KV1.3 expression and colocalization with CD8. (J) Isotype controls using nonspecific rabbit primary antibody followed by usual secondary label and with primary rabbit anti-human and nonspecific labeled mouse anti-rabbit secondary antibody showed no background staining.
Adapted from Reference #19, with the kind permission of Dr.Calabresi of the Pathogy Department, John Hopkins Hospital and theProc. Natl. Acad. Sci. USA.
Famial amyloidotic polyneuropathy (FAP) is a neurodegenerative disease that occurs in the PNS and is characterized by extracellular deposition of amyloid fibrils composed of misfolded transthyretin (TTR). The disruption of Ca2+ homeostasis by TTR thru N-type Ca2+ channels has been proposed as the mechanism for its cytotoxicity based on the finding that the specific blocker, Conotoxin GVIA significantly reduced the TTR-induced increased in internal Ca2+ concentration.14
Amyotrophic Lateral Sclerosis (ALS) is a chronic, progressive disease marked by gradual degeneration of the nerve cells in the central nervous system that control voluntary muscle movement. The disorder causes muscle weakness and atrophy. The cause is unknown, and there is no known cure. It has been found that the motoneurons which innervate tongue muscles are vulnerable to degeneration in ALS and this is linked to the differential expression of voltage activated Ca2+ channels.15 It was found that there is a 3.5 fold greater expression of P/Q type current in these motoneurons as opposed to those which ennervate extraocular muscles as was shown by immunolabelling using Anti-CACNA1A (CaV2.1) Antibody (#ACC-001). Identity of the channels was confirmed by determining the sensitivity of the Ca2+ currents to channel blockage using the N-type blocker ω-ContoxinGVIA and the P/Q blocker ω-Agatoxin-TK.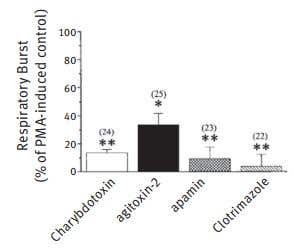
Figure 3. K+ Channel Blockers Reduce the PMA-Induced Respiratory Burst in Rat Microglia. Microglia were incubated in 2 mM DHR 123 and the fluorescence monitored for 5 min to ensure a stable baseline. A K+ channel blocker was perfused into the bath, the fluorescence recorded continuously for a further 5 min, then the bath was perfused with the blocker plus 100 nM PMA. The fluorescence signal from several cells in each field was monitored for a further 45 min, and the final average value compared with control cells from the same batches (number of cells from at least 3 rat litters indicated). The drug concentrations were 50 nM Charybdotoxin (#STC-325), 5 nM Agitoxin-2 (#STA-420), 1.2 nM Apamin, and 500 nM Clotrimazole. *P , 0.05 and **P , 0.001,values significantly different from controls.
Adapted from Ref. # 11, with the kind permission of Dr. L.C. Schlichter of the Toronto Western Hospital and the Am. J. Physiol. Cell. Physiol.
Multiple sclerosis (MS) is a chronic degenerative disease of the central nervous system in which gradual destruction of myelin occurs in patches throughout the brain or spinal cord (or both), interfering with the nerve pathways and causing muscular weakness, loss of coordination and speech and visual disturbances. MS is the most common neurological cause of disability in young adults in industrialized societies.16 Recent studies have identified changes in the expression pattern of specific Na+ channel isoforms as an important contributor to remission and progression in MS, and there is evidence suggesting that aberrantly expressed Na+ channels might also contribute to cerebellar dysfunction in MS.16 Changes that have, in the past, been attributed to the demyelination may, in fact, be heavily dependent upon the changes in ion channels.17 It has been suggested that KV3.1b interferes with conduction in demyelination axons.18 The presence of KV3.1b in CNS nodes has been shown using Anti-KV3.1b (KCNC1) Antibody (#APC-014). The expression of KV1.3 in postmortem MS brain tissue has been shown by immunohistochemical analysis and confocal microscopy using Anti-KV1.3b (KCNC1) Antibody.19 EAE is widely used experimental model for MS. In this model it has been shown that cell damage does not lead to an increase in endocannoinoid-mediated neuroprotection (as occurs in normal cells) because of disruption in the functionalilty of P2X7 receptors in the microglia caused by interferon-gamma. This was shown by immunoprecipitation experiments using Anti-P2X7 Receptor Antibody (#APR-004) in primary microglia cultures. These experiments showed that the overall expression of the receptors was not changed nor was their expression at the plasma membrane affected. It has been proposed that voltage gated Na+ channels can provide a route for Na+ influx into axons which triggers influx of damaging levels of intra-axonal Ca2+.20 The molecular identities of the channels involved has been investigated by immunocytochemistry using Anti-SCN2A (NaV1.2) Antibody (#ASC-002) and Anti-NaV1.6 (SCN8A) Antibody (#ASC-009) in the spinal columns of EAE mice. A significant increase in the number of dymelinated axons demonstrating NaV1.2 and NaV1.6 staining was shown. Shiverer mice have been widely used as a non-injury model in remyelination studies.21 The specific functional role of KV1.1 and KV1.2 in these mice was studied using Dendrotoxin-I (#D-390) and Dendrotoxin-K (#D-400). Distribution and changes in expression were studied using Anti-KV1.1 (KCNA1) Antibody (#APC-009) and Anti-KV1.2 (KCNA2) Antibody (#APC-010).
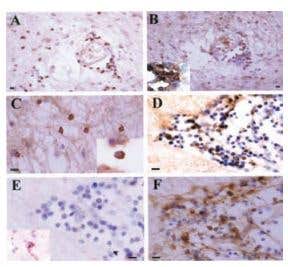
Figure 4. Expression of KV1.3 in Inflammatory Cells in Human MS Brain. Immunohistochemical staining of KV1.3 in inflammatory cells in human MS braining using Anti-KV1.3 (KCNA3) Antibody (#APC-002). Paraffin sections were stained by indirect immunoperoxidase for KV1.3, CD3, CD4, CCR7, and CCR5. Areas used in sectioning were from a white matter plaque. There were many perivascular inflammatory cells that stained positively for CD3 (A), KV1.3 (B), and CD4 (B Inset) on consecutive sections. (C) KV1.3 was also localized on inflammatory cells in the white matter parenchyma (Inset reveals membrane polarization of KV1.3 staining). (D) Consecutive sections through another perivascular infiltrate revealed numerous KV1.3- inflammatory cells, which were predominantly CCR7- (E) (Inset reveals rare CCR7 positive staining), and CCR5- (F). (Scale bar, 50 µm in A and B and 20 µm in C, D, E, and F.)
Adapted from Reference #19, with the kind permission of Dr.Calabresi of the Pathogy Department, John Hopkins Hospital and the Proc. Natl. Acad. Sci. U.S.A.
Parkinson’s diseaese (PD) is a slowly progressive degenerative disorder of the central nervous system characterized by slowness or poverty of movement (bradykinesia), rigidity, postural instability, and tremor primarily while at rest. Potassium channels have been implicated in the pathogenesis of PD. KATP channels comprised of Kir6.2 and Sur1 are abundantly expressed in substantia nigra dopamine neurons, which are the type of neurons that show degradation in Parkinson’s disease.22 GIRK-2 has been shown to be linked to the degeneration of dopaminergic neurons. Ventral mesencepalic cultures positive for GIRK-2, as shown by immunocytochemistry using Anti-GIRK2 (Kir3.2) Antibody (#APC-006) were more vulnerable to to MPP+ -induced neurotoxicity.23 The weaver mutation causes neuronal degradation and severe motor dysfunction caused by alterations in the inward rectifier channel and is often used as a model for PD. A population of granule cells expressed an inwardly-rectifying channel which was suggested to be GIRK-2 due to its sensitivity to QX-314 bromide (#Q-100).24
Duchenne muscular dystrophy (DMD) is a form of muscular dystrophy that is characterized by decreasing muscle mass and progressive loss of muscle function in male children. It is the most common congenital human neuromuscular disease. It is caused by a mutation in the dystrophin gene, a protein which forms a transmembrane complex linking the cytoskelton to extracellular proteins in many tissues. In dystrophin mutants immunostaining using Anti-GABA(A) α1 Receptor (extracellular) Antibody (#AGA-001) has shown that the size and number of GABA receptor clusters are decreased at cerebellar inhibitory synapses.25
Recently, mutations in ion channels have been shown to play a role in spinocerebellar ataxias. Spinocerebellar ataxia (SCA) is one of a group of genetic disorders characterized by slowly progressive incoordination of gait and is often associated with poor coordination of hands, speech, and eye movements. In SCA13, mutations in KV3.3 have been shown to have a key role and to alter the channel properties, leading to a change in the firing patterns of neurons, explaining the physiological manifestations of the disease.26 In a different type of ataxia, SCA63, a degenerative disorder of the cerebellum characterized by nearly selective and progressive death of Purknje cells, the mutation is in the α1A subunit of the neuronal P/Q-type voltage-gated calcium channel.27 This mutation leads to an expanded region of glutamine residues that shifts the voltage dependence of channel activation and rate of inactivation and impairs normal G-protein regulation of P/Q channels. Using the specific antibody Anti-CACNA1A (CaV2.1) Antibody has been shown that this pathological expansion can be expressed in multiple isoforms of the CaV2.1 subunit.
An important aspect of these findings is that the underlying mechanisms for many neurodegenerative diseases was thought to lie in accumulation of misfolded, aggregated proteins; the involvement of ion channels in neurodegenerative disease upons up a whole new avenue of theurapeutic possibilities.17







 protein")
