Voltage gated Na+ (NaV) channels mediate action potential genesis and propagation in most excitable cells. Other roles played by NaV channels in both excitable but also in non-excitable cells highlight their role as Na+ influx mediators in response to stimuli other than strong and brief depolarization of the cell’s membrane.
Introduction
The lipid membrane that surrounds the cell is impermeable to charged ions that are unequally distributed across it and this unequal distribution of charged particles generates polarization of the membrane potential (more negative inside). Ion channels are proteins that span the membrane and form channels through which these ions can pass and generate ionic electrical currents. Most channels exist in either an open or shut conformation and the transition between these two main conformations is highly controlled. Voltage sensitive channels respond to changes in membrane potential by transition to a conducting conformation1.
Voltage dependent Na+ (NaV) channels are sensitive molecular devices that respond to membrane depolarization with a transient Na+ influx2. This activity switches on the action potential (AP), the principal neuronal and muscular electrical signal1. This review focuses on NaV channels and their role in other cellular process ranging from neuronal modulation to cancer.
Structure, Nomenclature and Distribution of NaV Channels
NaV channels belong to the larger protein superfamily of voltage dependent channels that also include the KV and CaV channels3. The archetypical voltage sensing KV channel is formed by tetramerization of similar pore forming subunits and is also true for bacterial Na+ channels4. However, in higher organisms, NaV genes encode a protein with four similar domains, each corresponding to a single subunit in the KV channel tetramer3. Nine related genes encoding NaV channels have been identified in mammals, all forming one protein family (See Table)5. In addition, a structurally similar gene corresponding to a Na+ activated channel (NaV) which lacks the voltage sensor element, might form a second subfamily within NaV5. However this will not be discussed in this review.
NaV channels are expressed primarily in excitable tissues (nerve and muscle), with certain isoform-tissue/area specificity (See Table)5,6. This point is nicely illustrated by the detection of the cardiac isoform NaV1.5 in human embryonic stem cell derived cardiomyocytes7. However, recent findings demonstrate that neuronal NaV1.1 and NaV1.3 channels control the pacing of the mouse heart8,9 and that the cardiac isoform NaV1.5 is expressed in limbic regions of the rat brain10. In addition, several reports describe the expression of NaV channels in non-excitable cells11-13. This demonstrates that the tissue specificity of NaV channels is a broad observation, but important deviations from this pattern exist.
The nine human NaV channel genes are clustered on different chromosomes. Apparently structural, functional and pharmacological characteristics separates the channels encoded on chromosome 3 from all the others. This is manifested mainly by Tetrodotoxin (TTX) sensitivity demonstrated by all but the NaV1.5, 1.8 and 1.9 isoforms (See Table)5,6.

Reversible inhibition of NaV channels current in ND7-23 cells by 60 nM Tetrodotoxin citrate free (#T-500). Top: time course of inward current amplitude, recorded in a typical cell. Holding potential –100 mV, test pulse to –20 mV was delivered every 10 seconds. Period of TTX bath perfusion is indicated by the horizontal bar. Bottom: Traces before (blue) and during application of TTX.

CNS = central nervous system, PNS = peripheral nervous system.
NaV Channels as AP Initiators
Depolarization of the resting membrane potential is caused by the accumulation of positive charges on the inner interface of the plasma membrane. Thus, Na+ inflow results in membrane depolarization. This positively charged ion influx causes further depolarization of the membrane, which activates neighboring channels. This mechanism forms a positive feedback loop that lies at the heart of membrane excitability, as it forms the rising phase of most APs1. AP is a regenerative voltage spike, traveling along the cell membrane, by means of ionic currents and activation of voltage dependent ion channels (KV and KCa channels, co-localized with NaV, repolarize the cells’ membrane, terminating the signal). In neurons, AP travel rapidly for long distances along axons, transmitting cellular coded information.
Thus, most of the nine human genes encoding NaV channels participate in AP generation in neurons and muscle cells2,5. The combination of NaV channel isoforms expressed by an excitable cell determines parameters such as AP firing frequency, which are in the basis of neuronal coding. Indeed, different cell types express different combinations of NaV channels (See Table) and alterations in such expression patterns occur in different pathophysiological conditions. For example, changes in the levels of functional NaV channel expression forms the basis of changes in conduction properties of DRG neurons associated with chronic pain phenomena6. Recently, it was shown that the ubiquitin system participates in the regulation of NaV1.5 channel functional membrane expression in the heart14, adding another dimension to the regulation of cardiac excitability.
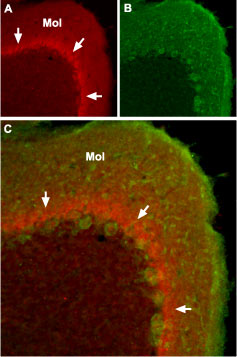
Immunohistochemical staining of NaV1.1 channel with Anti-SCN1A (NaV1.1) Antibody (#ASC-001) in mouse cerebellum. (A) The distribution of NaV1.1 (red) forms a band (arrows) in the molecular layer (Mol), close to the Purkinje cell bodies. (B) Purkinje nerve cells are stained with mouse anti Parvalbumin (green). (C) Confocal merge of NaV1.1 and Parvalbumin.
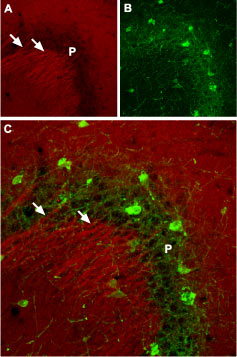
Immunohistochemical staining of NaV1.2 channel with Anti-SCN2A (NaV1.2) Antibody (#ASC-002) in mouse hippocampus. (A) NaV1.2 channel (red) in dendrites of pyramidal neurons in the CA3 region. (B) staining of Purkinje nerve cells with mouse anti Parvalbumin (green) demonstrates the restriction of NaV1.2 to dendrites (arrows) outside the pyramidal layer (P). (C) Confocal merge of NaV1.2 and Parvalbumin.

Immunohistochemical staining of NaV1.3 channel in rat brain using Anti-SCN3A (NaV1.3) Antibody (#ASC-004). NaV1.3 was visualized with immuno-peroxidase methods and final brown-black diaminobenzidine color product. There was strong staining of some axonal groups such as the mossy fibers in hippocampus (A) and cortico-striatal fibers (B).

1. Anti-SCN1A (NaV1.1) Antibody (#ASC-001) (1:200).2. Anti-SCN1A (NaV1.1) Antibody, preincubated with the negative control antigen.

1. Anti-SCN2A (NaV1.2) Antibody (#ASC-002) (1:200).2. Anti-SCN2A (NaV1.2) Antibody, preincubated with the negative control antigen.

1. Anti-SCN3A (NaV1.3) Antibody (#ASC-004) (1:200).2. Anti-SCn3A (NaV1.3) Antibody, preincubated with the negative control antigen.
NaV Channel Influxes Near the Resting Membrane Potential and in Non-Excitable Cells
Usually NaV channel influxes are brief and transient, generally activating within 1 ms after depolarization and terminating within 10 ms (while the stimulus is still on, a process called inactivation)5. Thus, AP arise from the brief activity of NaV channels, which respond to the extreme depolarization generated by AP. Nevertheless, several mechanisms and NaV channels may give rise to different activities, when the membrane voltage is at rest or during mild depolarization. This corresponds both to neuronal excitability and to the emerging role of NaV channels in non-excitable tissue physiology and pathophysiology. Both of these roles are briefly discussed below.
Neuronal modulation by NaV channels at rest and mild depolarization may arise from voltage dependent kinetic properties of channels such as inactivation15, persistence16 and resurgence17. These phenomena control the availability of NaV channels before and after an AP has passed, thus, influencing the probability of an AP to emerge18. NaV channel isoforms differ in the fine-tuning of their kinetic properties. Differences may arise from intrinsic properties, or from the ability to interact with other factors19. The expression patterns of the different isoforms gives rise to a large diversity of neurons with different AP firing behavior. Indeed, long lasting changes in expression patterns of NaV channels under pathological conditions for example, is strongly correlated with changes in such coding capabilities of neurons20.
Neuronal modulation may also arise from activation of channels by means other than voltage sensitivity. In mouse DRG neurons, NaV1.9 currents were found to be up-regulated by the inflammatory agent prostaglandin E2, in a G- protein dependent manner21. Another important example is the activation of the TTX-resistant NaV1.9 channel by neurotrophic factors (NT)22,23. This was shown in hippocampal neurons, where BDNF via TrkB receptors and NaV1.9 channels activated a long lasting Na+ current (about 200 ms) and a similar current was fourfold longer in neuroblastoma cells23. In frog sympathetic neurons, NGF upregulated both TTX-sensitive and TTX–resistant NaV currents24. On the other hand, NGF and GDNF reduced the NaV1.3 expression that accompanied DRG axotomy25. These interactions may contribute to pain sensation and pathophysiology, with differential specific interactions between sets of NTs and NaV channels6. Such interactions lie at the base of differential analgesic effects of different NTs, mediated by regulation of NaV channels in the peripheral nervous system25.
All these subthreshold activities may be the link and explanation for the expression of these highly voltage sensitive channels in non-excitable cells that do not fire AP.
Ion channel activity is an important factor in volume regulation and the associated motility of cells. A related mechanism is suggested for the ability to migrate, acquired by transformed glial cells, on their way to spread brain cancer26. Several NaV channels were reported to be upregulated in prostate cancer cell lines12,27,28. Interestingly, the magnitude of such NaV current was shown to be dependent on environmental factors that are present in the cell culture serum29. The role played by NaV channels, is suggested by the positive correlation between the rate of invasiveness and the level of NaV current or expression12,30. Most importantly, TTX, a highly specific (for NaV, but not very selective between the six sensitive isoforms), NaV channel blocker, reduced these cells’ invasiveness12. A possible role for NaV channels in volume regulation was demonstrated in Jurkat T cells, where Saxitoxin (STX) sensitive NaV channels play a role in apoptosis related cell shrinkage and death13,31. In addition, the NaV auxiliary subunit β3 (SCNB3) was found to be up-regulated in cancer cell lines in a p53 dependent manner, suggesting a role for NaV channels in induced apoptosis of cancer cells32.
These observations highlight the notion that voltage activated channels might be expressed in non-excitable cells, possibly under extreme cellular conditions.

Immunohistochemical staining of NaV1.5 channel with Anti-NaV1.5 (SCN5A) (493-511) Antibody (#ASC-005) in rat heart. Transversal section of the ventricular wall in the epicardium area also shows a big artery and myocardial area. DAB product is brown and counterstaining is Cresyl Violet. (A) Shows strong staining of myocytes (green arrow); no staining is evident in smooth muscle (red arrow), endothelium (black arrow), connective tissue (fibroblasts) (yellow arrow) or epicardial epithelium (blue arrows). (B) Enlargement of (A).

Immunohistochemical staining of NaV1.8 in adult rat dorsal root ganglion (DRG) with Anti-NaV1.8 (SCN10A) Antibody (#ASC-016). NaV1.8 Staining (red) was cytoplasmic and the intensity varied among DRG cells. There was a partial overlap in the distribution of NaV1.8 and neurofilament 200 (green).

Immunohistochemical staining of NaV1.9 channel in rat dorsal root ganglion (DRG) with Anti-SCN11A (NaV1.9) Antibody (#ASC-017). Cells within the DRG were stained (see solid line frame enlarged in (B)) as well as fibers and the area of entry of dorsal root into spinal cord (see dashed line frame enlarged in (C)). The counterstain in (B) and (C) is DAPI, a fluorescent dye visualized in the UV range.

1. Anti-SCN11A (NaV1.9) Antibody (#ASC-017) (1:200).2. Anti-SCN11A (NaV1.9) Antibody, preincubated with the negative control antigen.
NaV Channelopathies
Involvement of NaV channels in patho-physiological conditions may arise either from genetic alterations in channel activity or from changes in functional expression levels.
Mutations in coding for NaV channel proteins cause of several hereditary neuronal33, cardiac34 and muscular35 diseases. In addition, the mutation may occur de-novo (neither parent carries it) and such a mutant NaV1.5 channel was linked to Sudden Infant Death Syndrome36. In many of these mutations, the channel activity is up-regulated due to reduced inactivation, thus resulting in elevated baseline activity33.
Altered expression patterns and level of NaV channels in pathological states is demonstrated in Multiple Sclerosis (MS) and may contribute to specific phenotypes of this disease20. In addition, it was shown using specific antibodies that co-localization of NaV with the Na+-Ca2+ exchanger, plays a role in the increased axonal degradation seen in a mouse model of MS37. NaV channel expression is also changed at sites of axonal injury, conferring altered firing behavior to these injured neuronal branches6. Such a mechanism is the basis of abnormal pain phenomena as well.

Immunocytochemical staining of murine portal vein smooth muscle cells for NaV1.6 and NaV1.7 voltage-dependent Na+ channels. Fluorescence images of a single confocal plane of the cell labelled with (A) Anti-NaV1.6 (SCN8A) Antibody (#ASC-009); (B) Anti-NaV1.7 (SCN9A) Antibody (#ASC-008). (incubation with antigen peptide eliminated staining, data not shown). Calibration bar: 10 μm. (C) Summary data on intensity of fluorescence, expressed as average pixel fluorescence. The values of all pixels in the cell’s confocal plane were added up and then divided by the number of pixels (gray). The specificity of labeling was confirmed for both antibodies by greatly reduced fluorescence after pre-incubation with respective antigenic peptide (dark gray) or by virtual lack of fluorescence in the absence of primary antibodies (white).*: statistically significant.
Contributed by Vladimír Pucovský in association with Sohag Saleh, S.Y.M. Yeung, Sally Prestwich and Iain Greenwood, St. Geoge’s Hospital Medical School, London, UK.
Pharmacology of NaV Channels
NaV channels are involved in several different diseases and mediation of pain and indeed, these proteins are targets for many drugs ranging from anti-epileptic to local anesthetic agents5. In addition, many venomous organisms target their prey’s or attacker’s NaV channels38. In general, NaV pharmacophores are classified according to their receptor site on the channel. Among these, several molecules block the channel, while others support increased activity, either by enhancement of activation or by slowing inactivation5.
TTX, STX and μ-Conotoxins modulate NaV channels by blocking the pore from outside. TTX (See Table) and STX are largely non-selective between the different NaV isoforms5, while μ-Conotoxins are more specific and may differentiate between NaV current components39,40. These agents are widely used to block conduction in excitable tissue in a range of experimental procedures.
Local anesthetics and related compounds, such as QX-222 and QX-314, block NaV channels by binding to an intracellular site near the pore. These agents are capable of blocking all NaV isoforms when applied intracellularly. However, once applied from outside, it blocks channels differentially. This is probably correlated to the ability of these agents to penetrate the cell via the open channel41. Since these compounds can exert their blocking effect only once the channel has opened, they are called open channel blockers or activity dependent blockers.
Many venoms from different animals contain peptides that are NaV channel activators. Scorpion venoms are the largest source described so far38. Both α and β scorpion toxins bind to extracellular moieties on domain IV and II of the channel to slow inactivation or enhance activation kinetics, respectively5. Sea anemone NaV toxins such as Anthopleurin-C, APE 1-242 or ATX II43, 44, bind to the α scorpion toxin binding site and exert similar effects on inactivation4. Two small peptide toxins found in wasp venom, α and β-Pompilidotoxin, and promote NaV currents by slowing the inactivation process19. Such toxins are used to mimic, or specifically up-regulate, NaV subthreshold activities such as persistent and resurgent currents. Therefore, these NaV channels toxin activators might be used as powerful tools in NaV current induction at rest or in non-excitable cells.

Activation of NaV channels current in Native mouse NGF 2.5S protein (>95%)(#N-100) treated PC12 cells by 5 nM APE2-1. example current traces recorded in a typical cell before (blue) and during (black) application of Anthopleurin-C (APE-2-1) (#A-400). Holding potential –120 mV, test pulse to –20 mV was delivered every 10 seconds.
References
- Hille, B. (2001) Ion Channels in Excitable Membranes. 3rd edition.
- Catterall, W. A. (2000) Neuron 26, 13.
- Yu, F. H. and Catterall, W. A. (2004) Sci. STKE. 2004, re 15.
- Koishi, R. et al. (2004) J. Biol. Chem. 279, 9532.
- http://www.guidetoimmunopharmacology.org/immuno/index.jsp
- Ogata, N. and Oshini, Y. (2002) Jpn. J. Pharmacol. 88, 365.
- Satin, J. et al. (2004) J. Physiol. 559, 479.
- Maier, S. K. et al. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 3507.
- Lei, M. et al. (2004) J. Physiol. 559, 835.
- Hartmann, H. A. et al. (1999) Nature Neurosci. 2, 593.
- Fraser, S. P. et al. (2004) FEBS Lett. 569, 191.
- Bennett, E. S. et al. (2004) Pflugers. Arch. 447, 908.
- Bortner, C. D. and Cidlowski, J. A. (2003) J. Biol. Chem. 278, 39176.
- Van Bemmelen, M. X. et al. (2004) Circ. Res. 95, 284.
- Goldin, A. L. (2003) Curr. Opin. Neorobiol. 13, 284.
- Fleidervish, I. A. and Gutnick, M. J. (1996) J. Neurophysiol. 76, 2125.
- Afshari, F. S. et al. (2004) J. Neurohysiol. 92, 2831.
- Do, M. T. H. and Bean, B. P. (2003) Neuron 39, 109.
- Grieco, T. M. and Raman, I. M. (2004) J. Neurosci. 24, 35.
- Black, J. A. et al. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 11598.
- Rush, A. M. and Waxman, S. G. (2004) Brain Res. 1023, 264.
- Barde, Y. A. (2002) Nature 419, 683.
- Blum, R. et al. (2002) Nature 419, 687.
- Lei, S. et al. (2001) J. Neurophysiol. 86, 641.
- Leffler, A. et al. (2002) J. Neurophysiol. 88, 650.
- Sontheimer, H. (2003) Trends Neurosci. 26, 543.
- Bronstein-Sitton, N. (2004) Modulator 17, 4.
- Bogin, O. (2004) Modulator 18, 24.
- Ding, Y. and Djamjoz, M. B. (2004) Int. J. Biochem. Cell. Biol. 36, 1249.
- Grimes, J. A. et al. (1995) FEBS. Lett. 369, 290.
- Meir, A. (2004) Modulator 18, 2.
- Adaci, K. et al. (2004) Oncogene 23, 7791.
- Lossin, C. et al. (2002) Neuron 34, 877.
- Maraban, E. (2002) Nature 415, 213.
- Lehmann-Horn, F. and Jurkat-Rott, K. (1999) Phys. Rev. 79, 1317.
- Wedekind, H. et al. (2001) Circulation 104, 1158.
- Craner, M. J. et al. (2004) Brain 127, 294.
- Possani, L. D. et al. (1999) Eur. J. Biochem. 264, 287.
- West, P. J. et al. (2002) Biochemistry 41, 15388.
- Terlau, H. and Olivera, B. M. (2004) Phys. Rev. 84, 41.
- Sunami, A. et al. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 2326.
- Bruhn, T. et al. (2001) Toxicon 39, 693.
- Mantegazza, M. et al. (1998) J. Physiol. 507, 105.
- Mee, C. J. et al. (2004) J. Neurosci. 24, 8695.